Case 2: (read an already existing asy file)
Press "Read asymetric unit" button to select the data file
you want to use; a file selection box pops up:
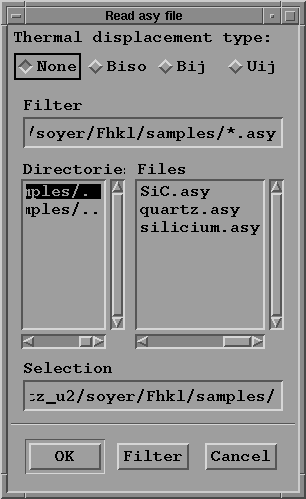
the program allows to take into account the thermal vibrations
provided in the xxx.asy file either as Bij, Uij anisotropic
thermal tensor componant or the B isotropic componant; the
choice is made by clicking on the correct toggle button
(in the case of quartz: "Uij"); then select the file named
"quartz.asy" by clicking on its name in the "Files" column.
Finally press the "OK" button to read the file, and to return
to "Generate cell" menu.
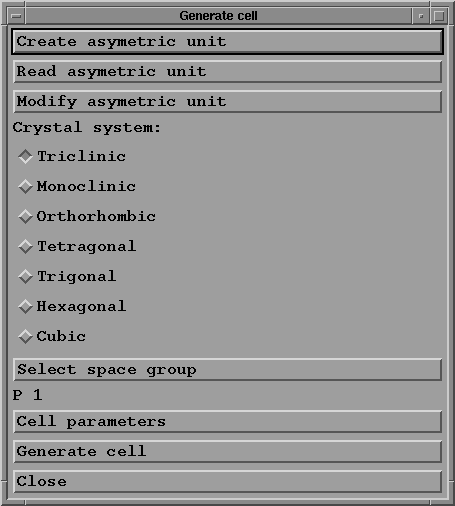
- Seven toggle buttons enable you to choose the crystal
system ("Trigonal" for quartz).
- The "Select space group" button gives you access to a
selection box:
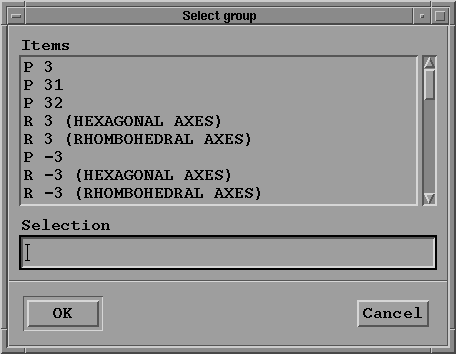
You may navigate using the scroll-bar on the right,
select the correct space group ("P 32 2 1" in the present
example), and click on the OK button to go back to the
previous menu.
- Press "Cell parameters" button to give the parameters and
angles of the unit cell:
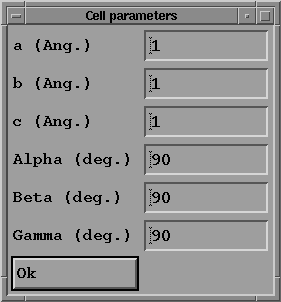
in the case of quartz these values are a = b = 4.9134, c = 5.4052,
alpha = beta = 90, gamma = 120. Click on the OK button
to go back to the previous menu.
- "Generate cell": when you press this button the program
creates the file xxx.cel (quartz.cel in the example),
which contains the description of all the atoms in the cell.
If an error occurs, a short error message is displayed
in a window, and other error information is printed
in the window from which Fhkl was executed.
Press "Close" to return to main menu.
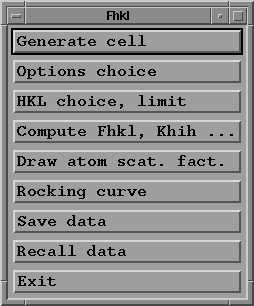
- Press "Option choice" button with your mouse;
a sub-menu window appears, where you may select the options
you want to use:
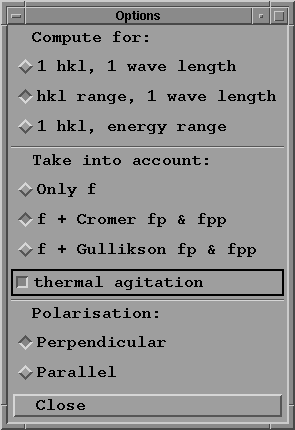
This menu offers the possible choices:
-to compute crystal characteristics for a given reflexion
at a given wave length;
-to compute crystal characteristics for all reflexions
in a given hkl range for one wave length;
-to compute crystal characteristics for one hkl and a range
of energy.
These computations take into account:
-the atomic scattering factors f as read from the file
named scat_fact.dat;
-optionally the anomalous dispersion corrections f' and
f'' (denoted fp and fpp in the program). These corrections
are either computed from a subroutine by D.T.Cromer, or
interpolated from data provided by E.M.Gullikson
(see references).
- Click, for instance, on "hkl range, 1 wave length" toggle
button;
- Click the right button to take into account f' and f''
computed with Cromer program, and thermal agitation.
- Choose the polarisation (this is taken into consideration
for the calculation of rocking curves only);
- Press "Close" to return to main menu.
- Press "HKL choice, limit" button with your mouse;
a sub-menu window appears:
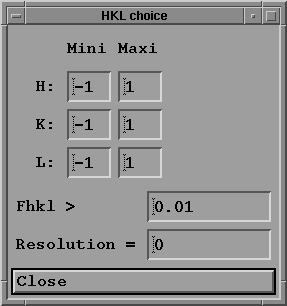
where you may:
- Indicate the hkl limit of the domain where you want to
compute crystal characteristics; choose for example
-1 1
0 0
0 0
- Ask the program to print results only for Fhkl greater than
a certain value (by default 0.01 to suppress extinction);
this option allows to discard small Fhkl values of no
interest.
- Limit the computation to a given resolution (in Ang.),
by default this resolution is 0 which means that all the
reciprocal space is explored.
- Press "Close" to return to main menu.
- Press "Compute Fhkl, Khih ..." button;
a sub-menu window appears:
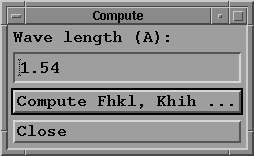
where you may:
- choose a wave length (in Angstrom): for example 1.54
(Cu Kalpha).
- press the "Compute Fhkl, Khih ..." button to begin the
computation.
During the execution, all the results are written
on the screen and in a file named Fhkl.log. Another
file named xxx.res (quartz.res for instance) is also
created. It is intended for use as entry to other
programs.
As a test, check that for the 100 reflexion the modulus
of Fhkl is approximately 16.3282.
Note that the look of this menu is not the same when using
the option "1 hkl, energy range", where you must provide
an energy step and maximum limits in eV.
- Press "Close" to return to main menu.
- Press "Draw atom scat. fact." button;
a sub-menu window appears:
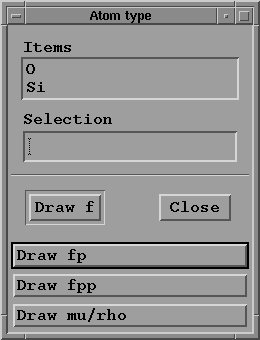
where you must select one of the possible atom types for
the studied material. Then you may draw:
- f versus sin(theta)/lambda
- f' versus energy
- f'' versus energy
- mu/rho versus energy
depending on the option previously chosen in the
"option choice" menu.
- Press "Close" to return to main menu.
If you now press the "Rocking curve" button, an error
message is displayed since in the present example you
have chosen in the "Options choice" menu "hkl range,
1 wave length".
To draw a rocking curve it is necessary to go back
to "Options choice" menu and select the "1 hkl,
1 wave length" toggle button.
You now have access to the "Rocking curve" menu:
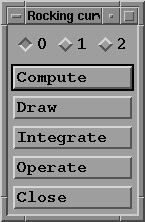
It is possible to store up to three curves labelled 0 to 2.
This number may be increased by modifying the MAX_RC
constant in the Fhkl.h header file and recompiling the
program.
- When clicked, the "Compute" button displays another
sub-menu:
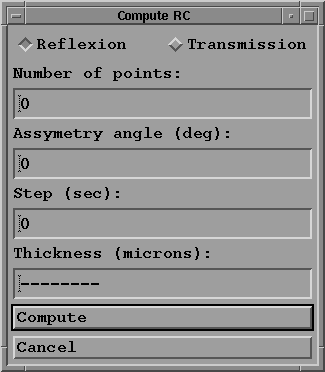
in which you:
- select the reflexion (or transmission) geometry;
- choose the number of points on the curve. It is recommended
to choose a power of 2 if later you want to compute the
convolution of two rocking curves. 256 is a good value.
- give the asymmetry angle in degrees if required;
- choose the step in seconds (for example 0.1);
- give the crystal thickness if in the transmission case;
- then compute the rocking curve.
At the end of the computation, the current menu automatically
closes.
You will now come back in the "Rocking curve menu",
where you may draw the rocking curve by pressing the
corresponding button:
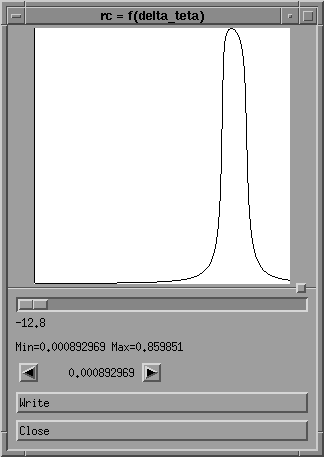
In this drawing curve menu, you may move a vertical cursor by
using the horizontal scroll-bar or by clicking on the left
or right arrows; each time you do this, the value of
delta theta under the scroll-bar and the value of the
reflecting power between the two arrows are updated.
You may also save the rocking curve in a file for further use,
by using the "write" button: this file will contain two
columns: the values of delta theta (in second) are indicated
in the first column, and the corresponding reflecting power
values in the second. You may for instance use it later
as data in your preferred drawing sofware (xprism2 for instance)
to obtain a more complete drawing with axes, titles ...
Press "Close" button to return to "rocking curve" menu.
- The "Integrate" button prints the value of the integral
of the current rocking curve.
- The "Operate" button gives access to a sub-menu
that permits various operations on rocking curves (R.C.).
For most operations there are a source rocking curve
and a destination rocking curve: this destination is
the current rocking curve as chosen in the "Rocking curve"
menu. So you must choose the destination R.C. before
entering the "Operate" sub-menu.
- Click on "Operate" button. A new sub-menu appears:
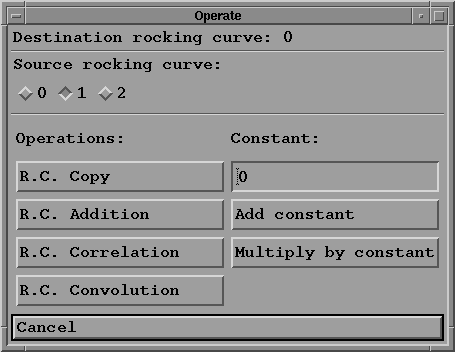
Possible operations are:
- select the source R.C. by clicking on one of the
toggle buttons (of course this R.C. must have been computed
first).
- copy the source R.C. into the destination R.C.: if the
destination is empty it is created and filled with the
source R.C., otherwise its content is replaced by the source R.C.
- add the source R.C. to the destination R.C.
- compute the correlation (in direct space) of the
destination R.C. by the source R.C.
- compute the convolution of the destination R.C. by the
source R.C.
Note that in all these binary operations the data stored in
the destination R.C. is lost and replaced by the result of
the operation.
- add a constant to the destination R.C.
- multiply the destination R.C. by a constant.
- do nothing by pressing the "cancel" button.
After the operation is completed, the current menu is
automatically closed.
- Press "Close" to return to main menu.
- The "Save data" button allows you to save the data entered
in the menus into a file (xxx.dat); the format of this file
is the same as the data file needed for Fhkline, and is
described later in this document.
- This may allow to recall the data by using the "Recall data"
button, in a further use of the program with the same material.
This drawing shows the different files used by Fhkl
(and Fhkline):
+-----------------+
.<------------------ ! cromer-orig.dat ! ------------------>.
! +-----------------+ !
! !
! +---------------+ !
! .<---------------- ! scat_fact.dat ! ---------------->. !
! ! +---------------+ ! !
! ! ! !
! ! +-----------+ ! !
! ! .<--------------- ! spgrp.gen ! --------------->. ! !
! ! ! +-----------+ ! ! !
! ! ! ! ! !
! ! ! +-----------+ ! ! !
! ! ! .<------------ ! xx.ASC ! ------------>. ! ! !
! ! ! ! +-----------+ ! ! ! !
! ! ! ! ! ! ! !
! ! ! ! +---------+ ! ! ! !
! ! ! ! .<---------- ! xxx.asy ! ---------->. ! ! ! !
! ! ! ! ! +---------+ ! ! ! ! !
! ! ! ! ! ! ! ! ! !
! ! ! ! ! ! ! ! ! !
+=============+ +=============+
! ! --------> +---------+ ! !
! Fhkl ! ! xxx.dat ! --------> ! Fhkline !
! ! <-------- +---------+ ! !
+=============+ +=============+
! ! +---------+ ! !
! '-----------> ! xxx.res ! <-----------' !
! +---------+ !
+----------+ +-------------+
! Fhkl.log ! ! Fhkline.log !
+----------+ +-------------+
cromer-orig.dat contains data for the calculation of the
dispersion corrections f' and f'' by Cromer program.
scat_fact.dat contains the scattering factors f for each
atom and ion from the International Tables for X-Ray
Crystallography.
spgrp.gen contains the generators for the 230 space groups.
The 92 files xx.ASC contain the data from E.M.Gullikson.
xxx.asy is the asymetric unit file given by the user.
xxx.dat is the second data file for Fhkline, and/or the save
file used by Fhkl.
The other files are the result files created by the program:
Fhkl.log is a log file that contains the trace and the
results of all operations performed by Fhkl.
Fhkline.log is the same for Fhkline.
xxx.res is intended for use as data with other programs;
it contains one line per reflection giving:
the indices h,k and l of the reflexion,
the interplane distance Dhkl,
the wavelength in Angstrom,
the structure factor (real part, imaginary part, modulus),
the dielectric susceptibility coefficient Khih (real part,
imaginary part),
the Khi_rh (real part, imaginary part),
the Khi_ih (real part, imaginary part)
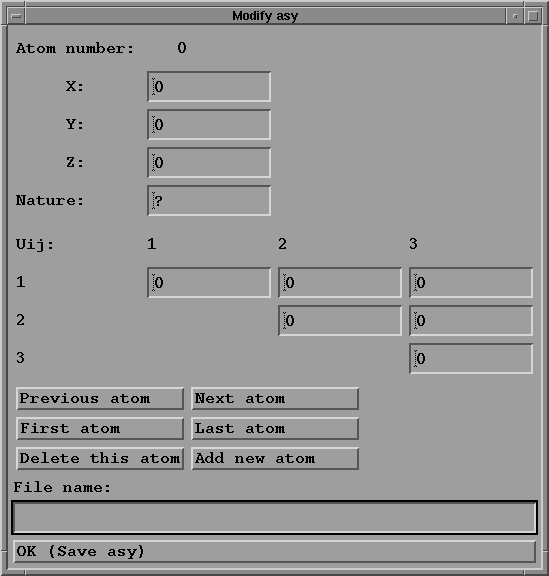
The xxx.asy file (see for instance quartz.asy) contains
information about atoms of the asymetric unit. One line
is written for each atom, and must contain:
-unit coordinates of the atom (3 real numbers): these
coordinates in the cell must be given with the origin
choice indicated in the International tables, volume A (1983).
-nature of the atom: a string of character (4 characters
maximum, for example Si for an atom of silicium or
Fe3+ for an iron ion); see the scat-fact.dat file for the
symbols for the elements.
-the (optional) thermal displacement components: B in the
isotropic case, or tensor components in the order
B11 B22 B33 B12 B13 B23, or U11 U22 U33 U12 U13 U23 in
the anisotropic case.
A second data file is required for Fhkline, which contains
the following lines:
1 - name of the file containing the asymetric unit
2 - crystal system:
0 = triclinic
1 = monoclinic
2 = orthorhombic
3 = tetragonal
4 = trigonal
5 = hexagonal
6 = cubic
3 - space group name (as in file spgrp.gen)
4 - cell parameters a, b, c (Ang.)
5 - cell angles alpha, beta, gama (deg.)
6 - thermal displacement type:
0 = none
1 = Bij
2 = Uij
3 = B isotropic
7 - option flag:
0 = one hkl, one wave length
1 = hkl range, one wave length
2 = one hkl, energy range
8 - polarisation flag:
0 = parallel
1 = perpendicular
9 - use fp and fpp in calculations:
0 = no
1 = use Cromer f' and f''
2 = use Gullikson f' and f''
10 - use thermal displacement in calculations:
0 = no
1 = yes
11 - Hmin, Hmax
12 - Kmin, Kmax
13 - Lmin, Lmax
14 - Fhkl minimum
15 - resolution (Ang.)
16 - lambda (Ang.) or energy step, energy minimum,
energy maximum (eV) depending on the option flag value
indicated in line 7.
Fhkl and Fhkline are mainly written in C language, but
also call some Fortran 77 subroutines: one to compute the f'
and f'' is due to D.T.Cromer, and the others for Fourier
transform come from A.Rimsky.
To install Fhkline on your workstation you need a C compiler,
a Fortran compiler and the corresponding libraries.
In addition Fhkl requires the X library (Xlib), the
X toolkit intrinsic library (Xt) and the OSF-Motif
library (Xm).
scat_fact.dat
cromer-orig.dat
spgrp.gen
in the directory sources:
Fgroup.h Fhkl.c
Fhkl.h Fhkline.c
Uij_to_Bij.c XasyCB.c
XatomCB.c XcellCB.c
XcompCB.c XexitCB.c
XhklCB.c XoptionCB.c
XrockCB.c XrrCB.c
XsaveCB.c XselectCB.c
Xstep1CB.c bosse.c
comp_rcCB.c cr_mat44.c
crft1.f cromer2.f
curve.c display_msg.c
do_rep.c fft.f
fft.h fortrc.f
gbosse.c gen_cell.c
gen_equiv.c get_asy.c
get_data.c get_gener.c
init.c oper_rcCB.c
rcft1.f save_data.c
trcfft.f wr_atoms.c
write_rcCB.c
makefile
in the directory doc:
Fhkl-doc.asci
Fhkl-doc.ps
in the directory samples:
quartz.asy
quartz.dat
in the directory gullikson:
xx.ASC (92 files)
A sample makefile is provided with the source files. The first
thing to do is to edit this makefile and to replace the
compiler names, options and library names by those for your
system, at the beginning of the file.
You must then edit the header file Fhkl.h to modify the
GEN_FILENAME, the SF_FILENAME, and the GULLIKSON_DIR:
these variables define the complete pathnames to access
datafiles provided with the program:
spgrp.gen that contains the generators for the 230 space
groups; scat_fact.dat that contains the scattering factors f
for the atoms and ions, from International Tables for X-Ray
Crystallography; the directory that contains the data from
Gullikson and al.
Also in cromer2.f you must modify DATAFILE on line 171.
this variable contain the complete name of the data file
needed for Cromer calculations (cromer-orig.dat).
The way to call Fortran functions from C is machine dependent:
on some computers it is necessary to add an underscore (_) at
the end of Fortran function names in the C calling programs. If
this is the case, define BLANC_SOULIGNE in the variable CFLAGS
in the makefile (as for example for Silicon-Graphics).
You may now run the makefile to compile and link Fhkl and
Fhkline.