BAXMAP v1.0
(Baxter’s Molecular Adjustment Program).
by
Ian Baxter
Imperial College of Science, Technology and Medicine, London.
Figure 1. BAXMAP.
Presentation:
BAXMAP is a PC program for visualisation , transformation and analysis of crystallographic coordinates.
BAXMAP allows for both crystallographic and non-crystallographic transformations, with such modifications as bond lengths, angles and torsions, as well as reorientations of molecules within the unit cell. Such facilities are normally only found within high-end molecular modelling packages.
BAXMAP interfaces with the freely available POV-Ray ray-tracing package to allow the production of professional quality ray-traced presentations.
To retrieve Baxmap : ftp://ftp.lmcp.jussieu.fr/pub/sincris/software/graphic/Baxmap.zip
Acknowledgments.
BAXMAP is written in 100% ANSI C, compiled with D. .J. Delorie’s port of the GNU C compiler (see http://www.delorie.com). PC specific graphics and GUI use the Allegro library by S. Hargreaves (see http://www.talula.demon.co.uk/allegro/). The program runs in 32 bit protected mode, under DOS using the DPMI server CWSDPMI by C.W. Sandman. POV-Ray is copyright 1991, 1997 by the POV-Ray team (see http://www.povray.org). BAXMAP is currently only supported under DOS, but although untested, should run under a dos box in windows95.
BAXMAP comes with absolutely no warranty. Use of the program is entirely at the users own risk, the author will not be held responsible for any consequences, arising directly or indirectly, from use of the program.
Any comments, suggestions for improvements or general gossip can be emailed to the author at:
baxterian@hotmail.com
Please let me know what you think of BAXMAP, and thank you for taking the time to try it.
Introduction.
BAXMAP provides interactive visualisation, analysis and alterations of molecular geometry. The program grew out of our interest in modelling polymer systems. We needed a simple tool which enabled us to take a molecular fragment, allow us to edit the fragment, resize its unit cell and to reorientate the fragment within this cell. Due to the non-crystallographic nature of these reorientations, no cheaply available crystallographic packages were available to perform these transformations, thus BAXMAP was born. BAXMAP has been under constant evolution. Initially the core functions worked only in text mode. Later a GUI was built around this core functionality, allowing realtime rotations and selection of atoms for editting. When it became necessary to produce presentation quality output the POV-Ray interface to was added, which is more or less were we are today. BAXMAP continues to evolve, with further features being added as and when we require them.
In it’s simplest form, BAXMAP can be used for viewing SHELX type files. It can also be used as an interface between SHELX results and input files - allowing atoms to be assigned to Q-peaks. At it’s most advanced BAXMAP allows for non-crystallographic manipulations including alteration of bond lengths, angles and torsions, resizing of the unit cell and realigning molecules within this cell. Connectivity information, which is automatically calculated when a SHELX file is read, can be editted.. Individual atoms or groups of atoms can be selected to allow BAXMAP commands to work only on certain atoms. The current state of BAXMAP, including the array of atoms and connectivity, the current view, selected atoms, labels, cell etc. can be saved to a BAXMAP format file
A POV-Ray interface has been incorporated to allow high-quality true-colour pictures to be output. The default POV-ray scene produced will render without modification, but by editting the file outstanding results can be produced.
The Baxmap Graphical User Interface (GUI) is shown in figure 2. There are basically 4 parts to the GUI;
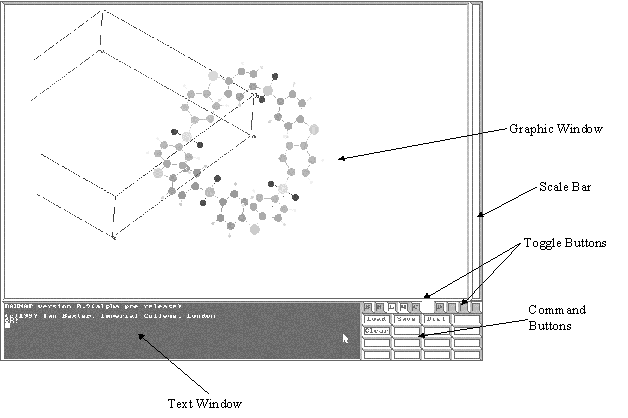
Figure 2. The BAXMAP graphical user interface.
The Graphics window allows real time rotation, translation and scaling of the current molecule. With the mouse ponter in this window, clicking left left mouse button and dragging the pointer rotates the molecule about the x and y screen axes; with the right button rotation is about the z screen axis. Double clicking the left mouse button whilst the pointer is over a atom will toggle the selection of said atom. Selected atoms are highlighted and labeled in red. A number of commands (see later) can act on individual, selected or all atoms.
Whenever a command is processed within BAXMAP the Graphic Window is automatically updated allowing interactive manipulation of the molecule.
The Text Window is the heart of Baxmap. ALL input and output to and from the program is performed through this window. Commands are typed directly to this window and all output is directed to this window. The Command Buttons send a predefined command to the Text Window.
BAXMAP Commands.
INPUT/OUTPUT commands:
Currently BAXMAP will only read/write SHELX type files. Where filename is required, if %F is used then a file selector dialog is presented to the user to enter a filename. Atoms can be inserted by directly inputting them to the text window, in the format
label sfac fx fy fz
or
label fx fy fz in which case Z is extracted from the atom label.
Load filename
Reads BAXMAP format file. BAXMAP format restores state BAXMAP was in when the program was saved.
Save filename
Save BAXMAP format file. Baxmap format saves full atomic coordinates, connectivity, cell information, selected atoms, and display options currently set in BAXMAP.
LoadShelx filename
Reads atoms from file filename into BAXMAP. This file can be a SHELX file, or can also contain BAXMAP commands. SHELX files are read until the end of file, so as to include Q Peaks (i.e. The END instruction is totally ignored!).
SaveShelx filename
Writes current atom list to filename in SHELX format. Please note that this alpha version of the program DOES NOT remenber sof, Uij’s, etc and so dummy values are inserted!. Also note that that the SHELX commands are also not remembered.
Delete [at1 at2 ... atn]
Removes atoms from the current structure.
Info [at1 at2 ... atn]
List a summary of the atom parameters to the Text Box. Atomic co-ordinates are in fractional co-ordiantes. If no atoms are specified, information is displayed for all atoms in current atom list.
List [at1 at2 ... atn]
Similar to Info, except atomic co-ordinates are orthogonal.
Scale [value]
Sets the current value of the display scale to value. The value of scale can also be adjusted interactivly using the scale adjustor box located next to the graphical display (see section ?.?). The keyword scale on it’s own displays the current value of scale in the Text Box (section ?.?).
Bind at1 at2
Adds bond between at1 and at2 to the connectivity array.
Free at1 at2
Removes bond from between at1 and at2 from the connectivity array.
Dist at1 at2 [value]
Calculates the distance between at1 and at2 in Å. If value is a included, this bond length is adjusted to this value. [ Please note, bond length adjustment is not really valid if bond is part of a ring system. If you do use this for a bond in a ring system, don’t blame me for the consequencies...]
Angle at1 at2 at3 [value]
Calculates the angle between at1, at2 and at3 in ° . If value is included, this angle is adjusted to this value. [ Not valid for ring systems, be warned...].
Torsion at1 at2 at3 at4 [value]
Calculates the torsional angle between at1, at2, at3 and at4 in ° . If value is included, this torsion is adjusted to this value.
Wind at1 at2 angle
The fragment bond about at1 and at2 is rotated by angle ° .
Cell [l ][a b c a b g ]
Cell prints the current cell info. If a b c a b g are included then the cell is adjusted to these retaining the current molecular geometry. l is included for compatability with the shelx cell instruction.
Align fx fy fz at1 at2
Aligns the vector between at1 and at2 with the vector specified by the fractional indices fx, fy and fz. For example, if one wished to align the current atom list with the C1-C2 bond parallel to the C axis, one would use the command Align 0 0 1 C1 C2. Please note that no translation of the atom list is performed; if you wish to overlay the two vectors you mush follow the align command with a push command (see below).
Push fx fy fz
Push at1
The push command can take two forms. If fx, fy and fz are specied the the current atom list is translated by theses fractional co-ordinates. If however at1 is specified, then the atom list is translated so as this atom is on the origin.
Select [at1 at2 ... atn]
at1, at2 to atn are added to the selection list. If the atoms are already in this list then they are removed from the list.
Mol at1
Selects the molecule containing at1.
Fragment at1 at2
Selects the fragment bonnected to the bond at1 - at2. Note, if you try to use this on part of a ring, or if at1 and at2 are not connected strange things may happen! Should really only be used for terminal fragments.
Clear
Clears the selection list. Can also be achieved via the command Select %S, but is included for historical reasons.
Interfacing with POV-Ray.
BAXMAP provides a basic interface to the program POV-Ray to allow ray-traced rendered presentations of the molecule. The POV-Ray instruction files produced by the program are complete in that the scene produced will render without modification to the file. However the default scene produced by BAXMAP may not represent the presentation to its best. For this reason I’ll include a few notes on editing the POV-Ray scene file to produce better representations. Below shows an example header as produced by BAXMAP’s POV instruction. The ellipses (…) represnts where the atom and bond information for the molecule is located.
//POV-Ray Scene file created by BAXMAP version 1.0 Alpha test release, Mar 24 1998
#include "shapes.inc"
#include "colors.inc"
global_settings { assumed_gamma 2.2 }
#declare atom_size=0.5
#declare bond_size=0.1
#declare dist= 91.57
//################ P Atom information ################
#declare P_radius= 1.10
#declare P_colour = Med_Purple
#declare P_finish = finish {
//ambient 0.1
//brilliance 3
//diffuse .1
//metallic
phong 0.8
//specular 1
//roughness .01
//reflection .75
}
.
.
.
// repeated for each atom type
.
.
.
camera {
location < 0.00, 0.00, dist >
look_at < 0.00, 0.00, 0.00 >
angle 20.000000
}
light_source {
< 0.00, 0.00, dist >
color rgb < 1.00, 1.00, 1.00>
}
background { color White }
union {
.
.
.
// molecule information
.
.
.
rotate <0, 0, 0>
}
A few pointers to note here. If the representation does not fill enough of the view, or overfills the view when rendered, then either decrease or increase the dist variable respectively. The atom_size variable is the percentage of the atomic radii at which the spheres for atoms are drawn. The bond_size variable is the radius of the bonds to be drawn (this example is for a ball and stick representation – the space fill represntation does not have any bonds, and therefore does not have a bond_size variable). The At_radius (where At = atomic symbol) variables are the default radii of the atoms, as used by BAXMAP. These can be altered to produce different representations as required. The At_colour variables are default colours for the atoms. These are assigned values by BAXMAP from the included pov header <colors.inc>, but they can equally be assigned pov rgb<x,y,z> values (see POV-Ray documentation for more details). You may find the colours are too light or too dark; these can be altered by multiplying the values by a scaler less than 1.0 to darken or greater than 1.0 to lighten the colour. For example, I often find that the default white colour used for protons by the program is too grey for my liking, and so replace the H_colour variable to be:
#declare H_colour = White * 1.4
You may find the actual orientation of the view is not quite how you would like it. Fine tuning of the orientation can be achieved by altering the rotate instruction at the end of the file.
By altering these variables and using the following commands within BAXMAP stunning publication quality presentations can be achived.
Summary of BAXMAP POV-Ray Interface Commands.
POV [filename]
Writes a pov-ray input file named filename (default Baxmap.pov), before lanching POVRAY. If for any reason povray can not be lauched, control is passed to BAXMAP, else POVRAY continues rendering the image. Once completed POVRAY waits for a keypress before handing control back to BAXMAP.
POVColor colour at1 at2 ... atn
Changes the colour of atoms at1 at2 ... atn to color for use in povray. colour can be any string, whether supported by POVRAY, eg a colour from POVRAY’s included colour definitions (see POVRAY documentation), or a user specified string to be #declare’d by the user.
POVFinish finish at1 at2 ... atn
Changes the finish for atoms at1, at2. .. atn to finish. The finish doesn’t need to ba a valid finish, but if not a user defined #delcare instruction must be added to the produced POV-Ray scene file before the representation will render.
POVBackground colour
Sets the background colour used by the presentation to colour.
POVType [n]
PovType reports the current presentation style. Parameter n can be used to set the style to :
n = 1 : Ball and Stick represenatation
n = 2 : Spacefill representation
n = 3 : Stick representation
Last updated Y.E. 25/10/1999