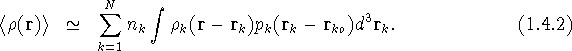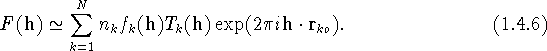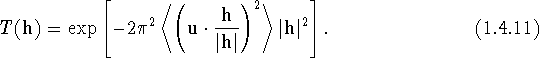The structure factor of reflection h is given in a fairly general form by the Fourier transform of the average density of scattering matter
with the integration extending over the repeating structural motif,
confined to
a single unit cell. The brackets denote a double averaging over the possible
displacements of the atoms from their mean positions - a time
average over the atomic vibrations in each cell, followed by a space average
that consists of projecting all the time-averaged cells onto one and dividing
by the number of cells, h is a diffraction vector obeying the
Laue equations, and ![]() is the static density of the motif,
consistent with
the instantaneous local configuration of the nuclei in a unit cell.
is the static density of the motif,
consistent with
the instantaneous local configuration of the nuclei in a unit cell.
To reduce the above general picture to what is used in conventional crystal structure analysis, we first assume that the average density of matter in (1.4.1) can be regarded as a superposition of averaged atomic densities. This so-called isolated-atom approximation is essentially equivalent to assuming independently displaced atoms, a fair initial approximation, although not generally valid. The average density of scattering matter at the point r in a unit cell can then be approximated as
Here N is the number of atoms in the unit cell,
![]() is the occupancy factor of the kth atom,
is the occupancy factor of the kth atom,
![]() is the density (electron
density for X-rays, or a delta function weighted with the scattering length
is the density (electron
density for X-rays, or a delta function weighted with the scattering length
![]() for neutrons) due to atom k at a point r when the
nucleus of atom k is at
for neutrons) due to atom k at a point r when the
nucleus of atom k is at ![]() , and
, and
![]() is the probability density function (pdf)
corresponding to the probability for having atom k displaced by
the vector (
is the probability density function (pdf)
corresponding to the probability for having atom k displaced by
the vector ( ![]() -
- ![]() ) from its reference position
) from its reference position
![]() in an average unit cell, which will be the mean position if
in an average unit cell, which will be the mean position if
![]() is sufficiently symmetrical. It is
important to remember that
the approximations in (1.4.2) include the assumption that atoms are
not deformable, by bonding or otherwise, even though at this stage the
static atomic electron density,
is sufficiently symmetrical. It is
important to remember that
the approximations in (1.4.2) include the assumption that atoms are
not deformable, by bonding or otherwise, even though at this stage the
static atomic electron density, ![]() , has not
been assumed to be spherically symmetrical.
, has not
been assumed to be spherically symmetrical.
If eq. (1.4.2) is now substituted into eq. (1.4.1), and the order of the summation and integration is interchanged, the structure factor becomes
with
If the substitutions ![]() and
and ![]() are made, the integral in (1.4.4) becomes
are made, the integral in (1.4.4) becomes
The inner integral in (1.4.5) has the form of a conventional
convolution of the density of atom k with the pdf for a displacement of this
atom from its mean position; the outer integral is a Fourier
transform of this convolution. This transform is multiplied by an exponential
that depends on the mean position, ![]() , of atom k.
, of atom k.
By the convolution theorem, the Fourier transform of a convolution equals the product of Fourier transforms of the functions involved. When this theorem is applied to the outer integral in (1.4.5), we obtain the conventional approximation for the structure factor of a Bragg reflection
If we let ![]() and (as before)
and (as before)
![]() , then in (1.4.6)
, then in (1.4.6)
is the scattering factor or form factor of atom k (for neutrons this
is replaced by the
scattering length ![]() ), and
), and
is the Fourier transform of the pdf, ![]() , for the displacement of the kth
atom from its reference position, r
, for the displacement of the kth
atom from its reference position, r ![]() . This term contains the
dependence
of the structure factor on atomic displacements, and has been known by the
names ``atomic Debye-Waller factor" and ``atomic temperature factor"
(see section 1.5). There are no restrictions on
the functional form of
the pdf in the integrand of (1.4.8).
. This term contains the
dependence
of the structure factor on atomic displacements, and has been known by the
names ``atomic Debye-Waller factor" and ``atomic temperature factor"
(see section 1.5). There are no restrictions on
the functional form of
the pdf in the integrand of (1.4.8).
Let us now recall that the structure factor equation used in routine refinement of atomic parameters is further simplified in two ways:
First, for X-rays, the static atomic electron density is assumed to have spherical symmetry. This reduces the atomic scattering factor to the form
which has been computed and extensively tabulated for all the neutral elements and many ions (Maslen, Fox & O'Keefe, 1992). The spherical-atom approximation necessarily removes fine details of the (calculated) electron density, but may be used routinely, and serve as a starting point for more refined determinations of atomic positions and studies of charge density (e.g., Coppens & Becker, 1992; Coppens, 1993).
Second, the pdf for atomic displacement is most frequently approximated by a
univariate or trivariate Gaussian, depending on whether the atomic
displacements are
assumed to be isotropic or anisotropic respectively. If a trivariate Gaussian
is assumed, and the atomic subscript k is omitted,
the resulting expression for ![]() from (1.4.8) is
from (1.4.8) is
Equation (1.4.10) can be derived from the theory of lattice dynamics in the
harmonic approximation, which considers only the (always present)
contribution of motion to the atomic displacement (e.g., Willis & Pryor,
1975). However, this equation may also be applied to static displacive
disorder. The form of the atomic Debye-Waller factor, ![]() ,
represented in (1.4.10) is the most common one in standard structure
refinements and will be discussed in Section 2.
Various other approximations
have been proposed for situations in which the Gaussian formalism is not
adequate, e.g., when the anharmonic contribution to the crystal dynamics
is significant; the most common are discussed in
Section 3.
,
represented in (1.4.10) is the most common one in standard structure
refinements and will be discussed in Section 2.
Various other approximations
have been proposed for situations in which the Gaussian formalism is not
adequate, e.g., when the anharmonic contribution to the crystal dynamics
is significant; the most common are discussed in
Section 3.
We present now a short discussion of common variants of eq. (1.4.10), which can be rewritten as
This shows that the exponent is proportional to minus the mean-square
projection of the atomic displacement u on the direction of the
diffraction vector h times the squared magnitude of h.
If we denote the projection of u on the direction of h by
![]() , and make use of the relation:
, and make use of the relation:
![]() , (1.4.11) becomes
, (1.4.11) becomes
As long as the atomic displacements are anisotropic, the value of the average
in (1.4.12) depends on the direction of h. This is then the
anisotropic Gaussian Debye-Waller factor, ![]() , which is discussed in
detail in Section 2. If, however, the atomic
displacements are isotropic, the
average in (1.4.12) is a constant determined by the structure alone, but
possibly different for non-equivalent atoms, and the left-hand side of this
equation no longer depends on the direction of h, but only on its
magnitude. This is then the atomic isotropic Gaussian Debye-Waller factor,
, which is discussed in
detail in Section 2. If, however, the atomic
displacements are isotropic, the
average in (1.4.12) is a constant determined by the structure alone, but
possibly different for non-equivalent atoms, and the left-hand side of this
equation no longer depends on the direction of h, but only on its
magnitude. This is then the atomic isotropic Gaussian Debye-Waller factor,
The lowest-order approximation to ![]() is the overall isotropic
Debye-Waller factor. It has the same form as (1.4.13), and presumes that
all the atoms have the same isotropic mean-square displacement,
is the overall isotropic
Debye-Waller factor. It has the same form as (1.4.13), and presumes that
all the atoms have the same isotropic mean-square displacement,
![]() . The whole crystal structure is assigned, in this
approximation, a single displacement parameter. This approximation is
used in initial stages of crystal structure determination by direct
methods.
. The whole crystal structure is assigned, in this
approximation, a single displacement parameter. This approximation is
used in initial stages of crystal structure determination by direct
methods.
We conclude this section with some remarks on the structure
factor for electron diffraction by a crystal. The density of scattering
matter, ![]() , is here interpreted as the distribution of electrostatic
potential within the unit cell. This potential is then approximated by a
superposition of electrostatic potentials contributed by individual
atoms, and the effects of motion are taken into account, as for
X-rays and neutrons, by the convolution of the potential of
an atom at rest with the probability density function describing the
atomic motion (e.g., Vainshtein and Zvyagin, 1993). The atomic
(spherical) scattering factor for electron diffraction,
, is here interpreted as the distribution of electrostatic
potential within the unit cell. This potential is then approximated by a
superposition of electrostatic potentials contributed by individual
atoms, and the effects of motion are taken into account, as for
X-rays and neutrons, by the convolution of the potential of
an atom at rest with the probability density function describing the
atomic motion (e.g., Vainshtein and Zvyagin, 1993). The atomic
(spherical) scattering factor for electron diffraction, ![]() , for an atom at rest and diffraction vector
, for an atom at rest and diffraction vector ![]() , is related to
that for X-rays by the Mott formula (e.g., Vainshtein, 1964) which
has the form
, is related to
that for X-rays by the Mott formula (e.g., Vainshtein, 1964) which
has the form
![]()
with ![]() the atomic number and
the atomic number and ![]() is the X-ray
form factor of atom k [see (1.4.9)]. This formula,
with the correct proportionality constants, has been used along with
other techniques in extensive tabulations of spherical form factors for
electron-diffraction (see, e.g., Cowley, 1992). The Debye-Waller
factor, here expressing the `smearing out' of the electrostatic potential,
is given by the same expression as that quoted above for X-rays and neutrons
(e.g., Vainshtein, 1964; Vainshtein & Zvyagin, 1993). The structure
factor for electron diffraction is therefore analogous to that
appearing in (1.4.6) but is often given in a different notation.
is the X-ray
form factor of atom k [see (1.4.9)]. This formula,
with the correct proportionality constants, has been used along with
other techniques in extensive tabulations of spherical form factors for
electron-diffraction (see, e.g., Cowley, 1992). The Debye-Waller
factor, here expressing the `smearing out' of the electrostatic potential,
is given by the same expression as that quoted above for X-rays and neutrons
(e.g., Vainshtein, 1964; Vainshtein & Zvyagin, 1993). The structure
factor for electron diffraction is therefore analogous to that
appearing in (1.4.6) but is often given in a different notation.