4.54. REGFE: Analyse errors
Authors: H. Wang, R.J. Barton and B.E. Robertson
Contact: Bev Robertson, Department of Chemistry, University of Regina, Regina, Saskatchewan, Canada S4S 0A2
REGFE calculates crystallographic functions and their estimated standard deviations (Busing et al., 1964). In the calculation of standard deviations, both the variances and the covariances of the parameters (Hamilton, 1964; Sands, 1982) are taken into account. REGFE may be used to create tables of bond lengths, bond angles and dihedral angles for publication and output them to the 'punch' file.
4.54.1. Introduction
REGFE calculates the values of derived quantities which are functions of the atomic coordinates and the crystal unit cell parameters and their associated standard deviations. These quantities are bond lengths, bond angles, dihedral angles, distances of atoms from planes and lines, and angles between planes and lines. The calculations of bond lengths, angles and dihedral angles of molecules or clusters can be performed, based on atom connectivity.
In least-squares structure refinement, the parameters of the crystal structure are not fully independent of each other; i.e., they are correlated. For any function of these parameters the covariances of the parameters should be included in the calculation of their standard deviation.
There are two variance-covariance (v-c) matrices which are involved in the calculation of the standard deviation; the matrix, C, of the cell parameters and the matrix, V, of the atom parameters. We assume that the cell parameters are fully independent of the atom parameters and the covariance terms involving a cell parameter and an atom parameter are set to zero.
The variance-covariance matrix of the atom parameters is derived from the inverse matrix of the normal matrix in least-squares refinement. If the weights for the observations (F, F2 or I) are correct, V is the inverse of the normal matrix. However, it is assumed that any deviations of the goodness-of-fit, S, from its expected value represent a scaling error in the variances of the observations, and thus can be corrected by multiplying V by S2; i.e.
V = A-1S2
where A is the normal matrix of the least-squares refinement, given by
Aij =  r[ w dFcal /
dpi dFcal / dpj ]
r[ w dFcal /
dpi dFcal / dpj ]
and S, the goodness-of-fit is given by
S2 = r [w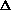 2]
/ (n-m)
2]
/ (n-m)
The symbols and notation in these equations have been defined in the CRYLSQ section.
The diagonal terms of the v-c matrix are the variances of the parameters and the off-diagonal terms are the covariances of the parameters. The correlation matrix, N, of the atom parameters is a normalized variance-covariance matrix. The elements of the correlation matrix are defined as:
Nij = Vij / [ Vii Vjj] (i,j = 1,2,3,...,m)
The correlation matrix of the atom parameters can be created and saved in the bdf cmx by entering the ms option code in CRYLSQ. Thus at least one cycle of least-squares refinement by CRYLSQ is required before running REGFE. If the correlation matrix from least-squares structural refinement is a blocked matrix, the parameters in different blocks are considered to be uncorrelated and the covariances of parameters in two different blocks will be set to zero. If the correlation matrix is not provided, only the variances of the parameters will be used and all the covariances of the parameters will be set to zero.
4.54.2. Standard Deviation Calculation
For a given crystallographic function of the atom parameters a1, a2, a3, ..., and cell parameters c1, c2, c3, ...
F = f(a1, a2, a3, ..., c1, c2, c3, ..., c6)
the variance (which is the square of the standard deviation, sigma) of the function can be obtained by
 2 = 2c + 2a
2 = 2c + 2a
The first term in the right hand side of the equation is the contribution to the variance of the function from the variances of the unit cell parameters:
2c = ij [ Cij dF/dcidF/dcj ]
and the second term is the contribution to the variance of the function from the variances of the atom parameters:
2a = ij [ Vij dF/daidF/daj ]
4.54.3. Atom Connectivity
The atom connectivity is established according to the atomic radii data. The symmetry operations of the space group may be applied for the establishment of the atom connection table. The number (1, 27, 125) of unit cells can also be specified for atom searching. The values of the atomic radii must be provided from the input bdf or atrad lines. The values of the atomic radii in the bdf can be modified by entering atrad lines. The min allowed radius is 0.015 A.
4.54.4. plane, line and atom Lines
In the function and error calculation, a plane can be represented by either a plane ID, which is predefined in a plane line, or by three atom IDs which define the plane. A line can be represented by either a line ID, which is predefined in a line line, or by two atom IDs which define the line. The equation of a plane can be created by entering the coefficients of the plane equation in either an orthogonal coordinate system or a fractional coordinate system, or by three atom IDs. Similarly, the equation of a line can be defined by entering the coefficients of a line equation in either an orthogonal coordinate system or a fractional coordinate system, or by two atom IDs. The forms of the equations of a plane and a line in factional coordinate system are given by:
Ax + By + Cz = D
and
(x- xo)/A = (y-yo)/ B = (z-zo)/C
respectively. However, when the coefficients of the equations are input on lines, only the variance of the coefficients will be used in the standard deviation calculations.
4.54.5. File Assignments
Optionally reads structure data from input archive bdf
Optionally reads correlation matrix from bdf cmx
Optionally outputs tables to line file pch
4.54.6. Examples
In this example, the atomic radii of Zn and Cl are modified and the bond lengths, bond angles, and dihedral angles of the molecule will be calculated based on the atom connectivity. The correlation matrix will be input from cmx and used in the standard deviation calculations.
reset psta 4 REGFE covar nlen nang lvc plane pln1 0 12.345 -8.456 3.004 5.842 plane pln2 0 12.462 3.145 8.593 -4.053 plane pln3 2 c01 c02 c03 plane pln4 2 c04 c05 c06 plane plna 1 0.6258 0.2449 0.7405 5.005 functn vol functn dis c01 c02 functn pap pln3 c04 c05 c06 functn dih cl01 zn01 o01 n01 functn ang c01 c02 c03 functn pda pln3 c04 functn pap c01 c02 c03 pln4 |
In this example, the calculation of the bond lengths, angles and dihedral angles is suppressed. The covariance of the atom parameter will be used in the standard deviation calculation and the variance-covariance matrix will be listed in the output.