2.2. System Terminology
2.2.1. compid: compound identification code
The compound identification code is a string of up to six alphanumeric characters entered as field 1 of the compid system command line. This code is usually referred to in program descriptions as the compound ID, or simply as the compid. The compid is inserted at the top of each printed page and used to construct the Xtal filenames. Note that if the compid is not defined its default value is XTAL. Users must be consistent about the definition of compid in order that the correct Xtal files will be opened (in other words, a compid line must always be entered, or never entered).
2.2.2. rcode: reflection status code
Reflection data in the Xtal system are assigned rcodes which indicate the reliability or condition of measurement. The rcode is a integer between 1 and 5.
measurement above the threshold of observability (e.g. > 3 sigma)
measurement below the threshold of observability (e.g. < 3 sigma)
measurement contains uncorrected systematic error
Friedel-related measurement is missing from a Friedel pair
measurement systematically absent for the space group in use
2.2.3. Atom labels
The rules for atom label construction are:
An atom label may be up to 24 characters in length.
An atom label may not contain imbedded blanks.
The alphabetic characters in the atom label are case-sensitive. Upper and lower case characters are preserved.
An atom label has up to eight sequential component codes numbered 0 to 7. Components 0 and 1 are concatenated; all other components are separated by an underline.
Component 0 is the atom type symbol which is entered on the CELCON line of STARTX. This symbol may be up to 8 chars in length. All characters are permitted, except digits '0' to '9' may only precede a '+' or a '-' character to indicate an oxidation state. Normally, but not necessarily, the element symbol is used so that STARTX can automatically assign scattering factors for the atom type. Typical atom type symbols are: C Cu S2- Cu&Fe Water
Component 1 is the atom sequence code. This may be any string of characters (excluding an underline) which must start with one or more digits. Some permitted sequence codes are: 1 3a1 23molA Examples of atom labels involving components 0 and 1 are: I C7 Cu2+4a C23molA 'C8%a' (this latter label will be plotted as C8
 - see below).
- see below).
The atom label construction is flexible, visually decipherable and well suited to computer applications. The components can be easily identified and stripped in a single pass from left to right along the label string. Note that the underline separators are only used if higher order components exist. If intermediate components are not needed they may be omitted provided the underline separators are inserted. For example the label C233__ggg is acceptable and decodes as component 0: 'C', 1: '233', 2: ' ' and 3: 'ggg'. There is no requirement that the same number of components be used in each label.
The plot routine PLOTX supports upper and lower case characters, Greek
characters, and superscripts and subscripts. Each Greek character is signaled
by a preceding percent % character. For example the digraph '%b' will
be plotted as '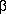 '; '%D' will be plotted as '
'; '%D' will be plotted as '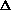 '. A
superscripted character is preceded by a caret ^, and a subscripted character
by a tilde '~'. Italic characters can be initiated by the vertical bar | character.
'. A
superscripted character is preceded by a caret ^, and a subscripted character
by a tilde '~'. Italic characters can be initiated by the vertical bar | character.
2.2.4. Frel, Fobs, Fcal: structure factor data
All structure factor data obtained by experimental measurement are entered into the bdf through various data entry programs. This data may be raw intensities from a diffractometer file, or values of F2 or |F| obtained by some previous data treatment. These data should consist of at least a unique asymmetric set of reflections.
For example, if the quantities supplied to ADDREF are intensities, various systematic factors may be applied to reduce this to the relative measured structure factor F(relative), or, as it is usually referred to in Xtal programs, Frel. When F2 is supplied, its square root is defined to be Frel. The observed (or measured) structure factor F(observed), or Fobs, is defined to be on the correct scale (e.g. in electrons) and is obtained from the product of Frel and the appropriate scale factor. Structure factor values which are calculated by the programs from atomic coordinates are referred to as F(calculated), or Fcal. The structure factor difference Fdif is obtained by subtracting Fcal from Fobs.
2.2.5. k: structure factor scale
The structure factor scales are used to place the values of Frel on the same scale as Fcal, as defined in the International Tables, Vol. I. That is, the scale factor is used to convert the value of Frel into Fobs.
The overall structure factor scale can be estimated with GENEV which uses the scattering factor and cell content information. The enot option can be entered to avoid placing E values on the binary file. In practice, these estimates have proved to be within 10% of the final scale factors for diffractometer data. In the case of film data, there may be several scale factors corresponding to data measured from different films. These scales are applied using scale group numbers.
These scale factors (whether overall, or for different groups of data) are usually refined by least squares calculations. During this refinement the quantity applied is the reciprocal of the scale factor, although all input is in terms of the actual Frel scale factor. Most programs which use the scale factors store and retrieve these quantities from the archive bdf. However, if needed, these values may be superseded by values from scale lines. All programs which calculate or refine scale factors will update the output bdf with the new values. The exception to this is the structure factor program FC and RSCAN.
In addition to the scale adjustments calculated by the least squares, programs
such as FC and RSCAN calculate the quantity k', the rescale factor
k'= hFcal /
hk Frel
hFcal /
hk Frel
If the scale k is correct, then k' will be 1.0. It should be noted that the practice of adjusting scale factors when not all atom data is known may be misleading. For example, a difference map for locating hydrogen atoms will tend to exhibit artificially lowered peaks because the scale factor does not yet contain the hydrogen atom contribution.
2.2.6. Atomic displacement parameters
A variety of atomic displacement parameters have been used in the crystallographic literature. Xtal uses the isotropic and anisotropic parameters U and Uij exclusively. The isotropic displacement factor in the structure factor expression is
t = exp-8* 2Us2
2Us2
The anisotropic displacement factor in the structure factor expression is
t = exp-2**2(U11h2a*2+......+2U23k l b*c*)