![[IUCr Home Page]](/iucr-top/logos/iucrhome.gif)
![[Commission Home Page]](/iucr-top/logos/cteach.gif)
![]()
![]()
![]()
Next: Use of the Photoelectric Absorption Measurements
Up: Anomalous Dispersion of X-rays in Crystallography
Previous: The Atomic Scattering Factor. The Oscillator
For the continuum of positive energy states, the summations become integrals.
The natural frequency for them changes continuously, so that rather than the
discrete value g(![]() ) it is necessary to define the oscillator density
(dg/d
) it is necessary to define the oscillator density
(dg/d![]() ) at the frequency
) at the frequency ![]() . The number of oscillators with
frequencies between
. The number of oscillators with
frequencies between ![]() and
and ![]() + d
+ d![]() is (dg/d
is (dg/d![]() )d
)d![]() . This number is zero for
. This number is zero for ![]() <
< ![]() , where
, where ![]() is
the frequency associated with the s absorption edge. The oscillator strength
due to all K electrons for instance, is obtained by integration in the whole
range of frequencies
is
the frequency associated with the s absorption edge. The oscillator strength
due to all K electrons for instance, is obtained by integration in the whole
range of frequencies ![]() to
to ![]() , i.e. the interval where the number
of oscillators related to the continuum of positive energy states is different
from zero:
, i.e. the interval where the number
of oscillators related to the continuum of positive energy states is different
from zero:
| (17) |
| (18) |
| (19) |
Bethe65 calculated the oscillator strengths g(s, m) for hydrogen-like atoms.
From equation (19), the generalized Thomas-Reiche-Kuhn rule can be obtained observing that: g(s, m) = -g(m, s). The justification of this argument is of a statistical nature, for both transitions should have the same probability, since their net result must average zero.
Thus,
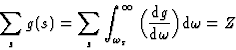 |
(20) |
![]()
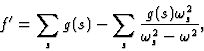
| (15') |
becomes
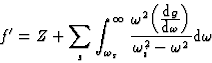 |
(21) |
![]()
Equation (21) only applies when ![]() , the incident frequency, corresponds
to a wavelength
, the incident frequency, corresponds
to a wavelength ![]() large in comparison with the atomic dimensions.
For frequencies higher than the natural frequencies of the atom and wavelengths
of the order of the atomic dimensions, we may substitute Z by f0, the normal
scattering factor, with good approximation and write:
large in comparison with the atomic dimensions.
For frequencies higher than the natural frequencies of the atom and wavelengths
of the order of the atomic dimensions, we may substitute Z by f0, the normal
scattering factor, with good approximation and write:
![]()
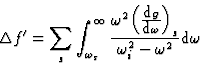 |
(22) |
Equations (21) and (22) are valid for any wavelength except for very short ones
when relativistic corrections are not negligible. Damping has also been
neglected: an approximation usually adopted in the calculation of
![]() and
and ![]() , which however does not
introduce unduly large errors except in the intervals
, which however does not
introduce unduly large errors except in the intervals
![]()
The `normal' scattering factor f0 has Z as a limiting value at low
frequencies for any angle of scattering and at very low angles for any
frequency. To obtain the real part of the dispersion correction one has to
integrate equation (22) so that the values of the oscillator densities
(dg/d![]() ) have to be calculated. This can be done from the atomic
wave-functions. Hönl62-63 made calculations for the K and L
electrons which
were assumed to be hydrogen-like. The result was quite satisfactory for the K
electrons but not for the L's. Later Hönl's method was applied by Eisenlohr
and Müller66 to the L electrons of several atoms.
) have to be calculated. This can be done from the atomic
wave-functions. Hönl62-63 made calculations for the K and L
electrons which
were assumed to be hydrogen-like. The result was quite satisfactory for the K
electrons but not for the L's. Later Hönl's method was applied by Eisenlohr
and Müller66 to the L electrons of several atoms.
The Hartree wave functions were tried by Cromer67 to obtain the oscillator strengths, but he found them inadequate for this purpose, particularly for the heavy elements. He tried then the relativistic wave functions without exchange, calculated by Cohen68 in the test cases of tungsten and uranium, finding better results. New relativistic wave functions computed by Lieberman, Waber and Cromer69 became available for all atoms, which included Slater's70- 71 approximate exchange correction and Latter's72 self-interaction term. Using these wave functions, Cromer67 calculated a set of oscillator strengths which have been in use up to now. From them, he obtained a set of dispersion corrections for elements 10 through 98 for five different wavelengths.
Copyright © 1981, 1998 International Union of Crystallography
IUCr Webmaster