![[IUCr Home Page]](/iucr-top/logos/iucrhome.gif)
![[Commission Home Page]](/iucr-top/logos/cteach.gif)
![]()
![]()
![]()
Next: The Imaginary Component
Up: Anomalous Dispersion of X-rays in Crystallography
Previous: The Oscillator Density
There is, in principle, a simpler approach to the calculation of the dispersion
terms by using the following relationship between the oscillator density
functions (dg/d![]() ) and the photoelectric absorption coefficient
) and the photoelectric absorption coefficient
![]() :61
:61
| (23) |
Using experimental values of ![]() (
(![]() ) one could obtain values for the
oscillator densities from equation (23). However, accurate values for
) one could obtain values for the
oscillator densities from equation (23). However, accurate values for
![]() (
(![]() ) are not presently available for all the elements in a useful
range of
) are not presently available for all the elements in a useful
range of ![]() . Then, the usefulness of an empirical method to obtain
(dg/d
. Then, the usefulness of an empirical method to obtain
(dg/d![]() )s, gs,
)s, gs, ![]() and
and
![]() based on the experimental values of
based on the experimental values of
![]() (
(![]() ) is very limited.
) is very limited.
A semi-empirical method has been used by taking the well known approximate
functional dependence of ![]() (
(![]() ):
):
| (24) |
By choosing the best experimental values for n and ![]() (
(![]() )integration of equation (17) should provide reasonably good values of gs.
One can substitute, in the general case, equations (23) and (24) in (17) to
obtain:
)integration of equation (17) should provide reasonably good values of gs.
One can substitute, in the general case, equations (23) and (24) in (17) to
obtain:
| (25) |
| (26) |
Equation (26) has been integrated in the general case by Parratt and
Hempstead.36 These authors have expressed their results in the form of
`universal anomalous dispersion curves', which are essentially the
representation of the integral in equation (26) with n as a parameter, as a
function of ![]() . When damping is neglected, these curves
are independent of the atomic number and of the electronic shell involved. To
obtain a particular value of
. When damping is neglected, these curves
are independent of the atomic number and of the electronic shell involved. To
obtain a particular value of ![]() , the value on the curve with
the correct value of n is multiplied by the oscillator strength calculated
from equation (25).
, the value on the curve with
the correct value of n is multiplied by the oscillator strength calculated
from equation (25). ![]() is then obtained by summation through
all s = K, L, M,
is then obtained by summation through
all s = K, L, M,![]() shells. The shape of the curves, reproduced in Fig.
1, is quite instructive. They are qualitatively correct and show that the
dispersion contributions from the various electron shells are rarely negligible,
or, in other words, as Parratt and Hempstead point out, there is practically no
region of normal dispersion.
shells. The shape of the curves, reproduced in Fig.
1, is quite instructive. They are qualitatively correct and show that the
dispersion contributions from the various electron shells are rarely negligible,
or, in other words, as Parratt and Hempstead point out, there is practically no
region of normal dispersion.
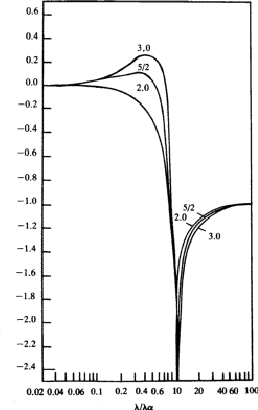 |
Since the method used by Parratt and Hempstead is based on experimentally determined values and uses exact integrations one should expect results in better agreement with the independent measurements made of the atomic scattering factor than is the case for the values obtained using Hönl's theory based on hydrogen-like electron shells. The calculations made by Parratt and Hempstead for the K region of copper and the L region of tungsten using only one term in the oscillator distribution for each electron shell, actually showed a less satisfactory agreement than Hönl's theory. This rather discouraging result was attributed by Parratt and Hempstead to (a) the difficulties inherent in the experimental measurements and (b) neglect of parts of the calculations in previous comparisons.
In fact, the experimental differences (f - f0) which they used to compare their calculations were presumably of a rather low precision, since they were based on values of f measured in the early 30's and presumably not very precise. The value of f0 subtracted in the iron case was the Thomas-Fermi f0 = 17.3 for plane (110) of Fe.
It would be interesting to remeasure the values of f using modern techniques and subtract better values of f0 as are currently available nowadays in order to make a definite comparison.
Copyright © 1981, 1998 International Union of Crystallography
IUCr Webmaster