4.2. ABSORB: Absorption corrections
Authors: George Davenport, Nick Spadaccini and James Stewart
Contact: Nick Spadaccini, Computer Science Department, University of Western Australia, Nedlands, WA 6907,Australia
ABSORB provides the following methods of correcting diffraction data for absorption effects: the Gaussian integration method devised by Busing and Levy (1957), the analytical method of de Meulenaer and Tompa (1977); corrections for spherical crystals; corrections for cylindrical crystals; and corrections derived from tables supplied by the user. Options include the choice of method, calculation of the mean path length Tbar, and the application of the absorption correction to data in the bdf.
4.2.1. Application
There are four steps in the determination of absorption corrections:
Careful measurement of the dimensions of the crystal.
Preparation of a description of the orientation of the crystal relative to its mounting on the diffractometer.
Determination, by recalculation, of the diffractometer angles and beam vectors for each reflection.
Calculation of the absorption correction for each reflection.
4.2.1.1. Analytical Method
In this method the convex polyhedral crystal is subdivided into tetrahedra. This method has the advantage that the absorption correction for a tetrahedron can be calculated analytically. The total absorption correction for the crystal is obtained by summation over all the tetrahedra which make up the polyhedron that is the crystal. The algorithm used is that given by Alcock (1974). For each tetrahedron the volume and transmittance are calculated.
4.2.1.2. Gaussian Method
In this method the convex polyhedral crystal is subdivided by an arbitrary grid set up inside the polyhedron. The path lengths of rays from the source to each point and out along the diffraction direction are calculated. A Gaussian integration over all the path lengths for all the grid points gives the absorption correction for each reflection. The methodology is essentially that of Busing and Levy (1957).
4.2.1.3. Spherical Method
Absorption corrections for spherical crystals of radius r are selected by entering sphere r on the ABSORB line. The values of A*, and T if requested, are derived from the algorithm of Tibballs (1982,1990) which is reportedly accurate to better than 1% for �r < 2.5.
4.2.1.4. Cylindrical Method
Absorption corrections for scattering in the equatorial planes of a cylindrical crystal of radius r are selected by entering cylinder r on the ABSORB line. Note that corrections for scattering from planes in the general orientation of a cylindrical specimen are not possible. The values of A*, and T if requested, are again derived from the algorithm of Tibballs (1982,1990) which is reportedly accurate to better than 1% for �r < 2.5.
4.2.1.5. Tables Method
Values of A* and T may be supplied by the user by selecting the table option on the ABSORB line. One or more astar lines containing values of the absorption corrections A* at intervals of 5� must follow the ABSORB line. If the tbar option is requested on the ABSORB line, one or more tbar lines containing values of T at intervals of 5� must also be included. Four point interpolation routines are used in both cases.
4.2.2. Crystal Descriptions
To specify the shape of a crystal, it is necessary to describe the faces of the crystal. The following three methods are available in the ABSORB program. Note that one and only one method may be used to describe the crystal.
4.2.2.1. faceml
Faces are described by giving, for each face, the Miller indices and the perpendicular distance from the face to an arbitrary origin within the crystal. The origin must be the same for all faces of the crystal. The Miller indices are transformed into the crystal Cartesian system using the B matrix (Busing and Levy, 1967), and the resulting vector is normalized, producing the normal to the face.
n = B h / |B h|
n1x + n2y + n3z = d (equation of face)
4.2.2.2. facept
Each face is defined by the coordinates of 3 points on the face in an arbitrary Cartesian system (it must be right-handed, orthogonal and normalized). The unit length in this system is 1.0 mm. The coordinate system origin must not be on an edge or face.
4.2.2.3. face
Each crystal face is specified by the 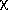 and
and  angles required to place
it in the diffracting position, plus the distance from the crystal face to an
arbitrary origin inside the specimen. This kind of measurement can be readily
done on diffractometers which have the viewing telescope mounted perpendicular
to the circle. The faces need have no relationship to the two orienting
reflections.
angles required to place
it in the diffracting position, plus the distance from the crystal face to an
arbitrary origin inside the specimen. This kind of measurement can be readily
done on diffractometers which have the viewing telescope mounted perpendicular
to the circle. The faces need have no relationship to the two orienting
reflections.
4.2.3. Diffractometer Description
The ABSORB program is designed to accept input data from any 4-circle
diffractometer, and to calculate absorption corrections for that
diffractometer. The motions of any diffractometer can be described by four
parameters; the direction of rotation of the 2 , , , and
, , , and
 circles. In the bisecting mode this reduces to 3-circles
(=). For
circles. In the bisecting mode this reduces to 3-circles
(=). For 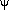 scans (rotations about the scattering vector) the
sense of rotation about the normal to the diffraction plane must be included.
Described below is the method which must be followed.
scans (rotations about the scattering vector) the
sense of rotation about the normal to the diffraction plane must be included.
Described below is the method which must be followed.
Set all circles to zero.
Look downward from above the diffractometer and note the directions of rotation of the 2
and circles as clockwise (C) or anti-clockwise (A).Look along the direction of the primary beam, from the x-ray source toward the goniometer head. Note the direction of rotation of the
circle as clockwise or anti-clockwise.Look downward from above the diffractometer and note the direction of rotation of the
circle as clockwise or anti-clockwise.Look down the scattering vector note the direction of motion during the
rotation as clockwise or anti-clockwise.
The following table lists the parameters for a number of diffractometer systems, both real and constructs used in papers. This information has been supplied by Jacek Grochowski.
| System | Senses of Rotation | ||||
| 2 | | | |
[a] | |
| Siemens (Syntex, Nicolet) | A | A | C | A | |
| CAD-4 | C | C | C | C | |
| Philips PW 1100 | A | C | A | A | C |
| Busing and Levy (1967) | C | C | C | C | C |
| Chidambaram (1980) | A | A | A | A | |
| International Tables, Vol. IV | C | A | A | A | A |
| Picker FACS-1 | C | A | A | C | |
| Picker with Finger refinement | C | C | C | C | |
| Notes: a. At the present time the ABSORB program does not handle any system where the sense of rotation is not uniquely definable. This occurs for the sense of rotation of for the
Syntex after Sparks. | |||||
The formulae of Busing and Levy can be modified using the equations which
follow (BL = Busing and Levy, C = clockwise, A = anti-clockwise). For any angle
 (, , , , ):
(, , , , ):
(BL) = (C) = -(A)
By introducing the parameters S(), S(), S(), S() and
S(), the senses of rotation of the circles of a diffractometer are defined
as:
S = +1 for clockwise rotation S = -1 for anticlockwise rotation
Three equations are produced which convert the angles used on a diffractometer, (DIFF), to equivalent angles in the Busing and Levy system.
(BL) = S() * (DIFF)
The program will assume the bisecting mode unless the angle
is stored on
the bdf (lrrefl:, IDN=1011). If, in the scan mode,
does
not fall within the diffractometer limits (specified in Fields 7-8 on the
diff line) the equivalent solution is used
= -, =  + , = +
+ , = +
4.2.3.1. Coordinate Systems
A number of different coordinate systems are used in determining absorption corrections. These are summarized as follows: (v(system) represents a column vector in a given coordinate system.)
The reciprocal lattice system may be non-Cartesian but is not necessarily. In this system a vector v(hkl) is represented as a linear combination of the right-handed reciprocal lattice vectors.
The crystal system is a right-handed Cartesian system obtained through the transformation of the hkl system v(crystal) = B v(hkl), where B is the transformation matrix defined using the direct and reciprocal lattice parameters. The B matrix is listed below.
Calculation of the absorption corrections is done using the coordinates of the crystal system.The
system is a right-handed Cartesian system which is rigidly
attached to the shaft of the diffractometer. The definition of the axes
is completely arbitrary. For two examples see Busing and Levy (1967) and
Coppens (1970). The ABSORB program uses the definition of Busing and Levy,
i.e., with all circles set to zero, the x-axis is along the scattering vector,
the y axis is in the direction of the primary beam, and the z-axis points
upward. The system is related to the crystal system by
the rotation matrix U as follows:v(
) = U v(crystal)
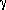
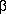

4.2.4. Plot Of Crystal Shape
Two views of the crystal shape are provided as options. These are a projection of the crystal shape along the three cell directions or a stereoscopic view of the crystal tilted 15� in z and -10� in y. These options are selected by entering proj or stereo on the ABSORB line. All plot information is stored on the plot bdf abs and the program PLOTX must be used to display the selected view on a graphical device.
4.2.5. File Assignments
Reads reflection data from the input archive bdf
Writes absorption corrections to the output archive bdf
Writes crystal shape commands to the plot bdf abs
Optionally, writes HKL lines to the line file pch
4.2.6. Examples
4.2.7. References
Alcock N.W. 1970. The Analytical Method for Absorption Correction.Crystallographic Computing. F.R Ahmed, S.R. Hall, and C.P. Huber, eds., Munksgaard. Copenhagen: 271-278.
Alcock, N.W. et al. 1972. An Improvement in the Algorithm for Absorption Correction by the Analytical Method. Acta Cyst. A28, 440-444.
Alcock, N.W. 1974. Absorption and Extinction Corrections: Calculation Methods and Standard Tests. Acta Cryst. A30, 332-335.
Busing, W.R. and Levy, H.A. 1957. High-Speed Computation of the Absorption Correction for Single Crystal Diffraction Measurements. Acta Cryst.10,180-182.
Busing, W.R. and Levy, H.A. 1967. Angle Calculations for 3- and 4- Circle X-ray and Neutron Diffractometers. Acta Cryst. 22, 457-464.
Cahen D. and Ibers, J.A. 1972. Absorption Corrections: Procedures for Checking Crystal Shape , Crystal Orientation, and Computer Absorption Programs.J.Appl. Cryst. 5, 298-299.
Chidambaram, R. 1980. Absorption Corrections for Single-Crystal X-ray and Neutron Data. Computing in Crystallography. R. Diamond, S. Ramaseshan, and K. Venkatesan, eds., Indian Academy of Sciences. Bangalore, India: 2.01.
Coppens, P. 1970. The Evaluation of Absorption and Extinction in Single-Crystal Structure Analysis. Crystallographic Computing. F.R. Ahmed, S.R. Hall, and C.P. Huber, eds., Munksgaard. Copenhagen: 255.
Coppens, P. & Hamilton, W.C. 1970. Anisotropic Extinction Corrections in the Zachariasen Approximation. Acta Cryst. A26, 71-83.
de Meulenaer, J. and Tompa, H. 1965. The Absorption Correction in Crystal Structure Analysis . Acta Cryst.19, 1014-1018.
Flack, H.D. and Vincent, M.G. 1980. Absorption and Extinction Corrections: Standard Tests. Acta Cryst. A36, 682-686.
Gabe, E.J. 1980. Diffractometer Control with Minicomputers. Computing in Crystallography. R. Diamond, S. Ramaseshan, and K. Venkatesan, eds., The Indian Academy of Sciences. Bangalore, India: 1.01.
Grochowski, J. Personal communication.
Tibballs, J.E. 1982 The Rapid Computation of Mean Path Lengths for Spheres and Cylinders. Acta Cryst. A38, 161-163.
Tibballs, J.E. Personal communication.