4.60. SCATOM: Calculate pseudo-atom form factor.

Authors: Doug du Boulay
Contact: Doug du Boulay, Materials and Structures Laboratory, Tokyo Institute of Technology, Nagatsuta, Midori Ku, Yokohama, Japan.
SCATOM takes X-ray scattering structure factors from a single element crystal structure and deconvolutes them into the elemental form factor on a grid of stol values.
4.60.1. Overview
SCATOM was written to try and obtain form factors accounting for the valence density of neutral, noninteracting atoms. Those form factors could allow the calculation of general theoretical valence density structure factors and thereby difference valence density maps, which highlight the changes to the calculated valence density arising from chemical interactions. Such maps aught to be comparable to experimentally determined difference (total) density maps.
As yet, it hasn't proven so useful.
4.60.2. Purpose
Abinitio density functional calculations of valence electron density using plane wave solutions and pseudo-potential approximations are frequently seen in the literature. These valence densities are not experimental observables in elastic X-ray scattering experiments because the X-rays are scattered by the total electron density. It is therefore quite useful, for comparative purposes, if the valence densities can be transformed to difference valence densities for direct comparison with experimental charge density results.
The difference valence density can be calculated iether by subtracting the standard crystallographic promolecular density [1] from the theoretically calculated total density directly in real space, or alternatively by Fourier transforming the reciprocal space structure factor differences. Being a crystallographic data analysis program Xtal is well suited to the the latter option.
There are two different approaches to calculating the difference density. One is to add the neglected core electron density contribution back to the theoretical valence density and then subtract the promolecular density from the total theoretical density, in the manner of a standard crystallographic difference density analysis. The second approach, which this program could assist towards, is to use the theoretical valence density of a lattice of superposed but noninteracting equivalent neutral pseudo-atoms, i.e. a valence density analogue of the promolecule, and subtract that from the compound whose valence density was actually being calculated, iether in real space or Fourier transfromed from reciprocal space.
In the former approach there is some degree of uncertainty as to exactly what form the core density should take. Generally it will resemble that of an inert gas, depending strongly on the pseudo-potentials actually used. However, the radial density distribution could differ markedly from that approximation, and it is probably not easily characterised.
The alternate approach is to use exactly the same machinary used to calculate
the abinitio valence density being studied, to calculate individually,
the spherically symmetric neutral pseudo-atom valence densities to be
subtracted from those in those actually calculated abinitially.
If the valence density of a single isolated atom is reverse Fourier transformed
in to structure factors, then this program SCATOM can
calculate the sin /
/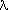 form factor
curve of a single pseudo-atom required to reconstruct the promolecular-valence
density of the extended lattice.
form factor
curve of a single pseudo-atom required to reconstruct the promolecular-valence
density of the extended lattice.
4.60.4. Examples
Calculate the pseudo-atom form factor of Si
compid si : using a silicon lattice MAPXCH abi2map noterm abifile si_o_DEN : read ABINIT map UOV 0.22 : The file si_o_DEN contains the spherically : symmetric non-interacting neutral atom : valence density calculated based on a : Troullier-Martins pseudo-potential ADDATM upd UOV 0.0000 : force to zero for SCATOM ADDREF : Generate dummy reflection data reduce nocon :limits *9 yes : for whole sphere - assuming MAPXCH wavelength? hklgen hkl frel sigf SORTRF aver 1 frel : Remove redundant reflections RFOURR th 0.000001 : Reverse fourier transform valence density idnums $1 304 801 802 700 MODHKL : Purge unphased reflections purge -4.+19 $1 1 1801 SCATOM ove : Calculate Si-pseudo-atom form factor. finish |