Following the C&EN article “Duplicate structures haunt crystallography databases”[1] in December 2025, the IUCr newsletter in February 2026 featured the article “Duplicate Dilemma – when should experimental crystallographic databases be concerned about near-duplicate crystal structures?”[2].
Though the words “crystal structure” appeared in the latter article[2] more than 10 times, no definition was given. The IUCr newsletter in April 2021 included the article “Change to the definition of 'crystal' in the IUCr Online Dictionary of Crystallography” (https://www.iucr.org/news/newsletter/volume-29/number-2/change-to-the-definition-of-crystal-in-the-iucr-online-dictionary-of-crystallography), which finished by saying that “a real-space definition mentioning periodicity is needed”.
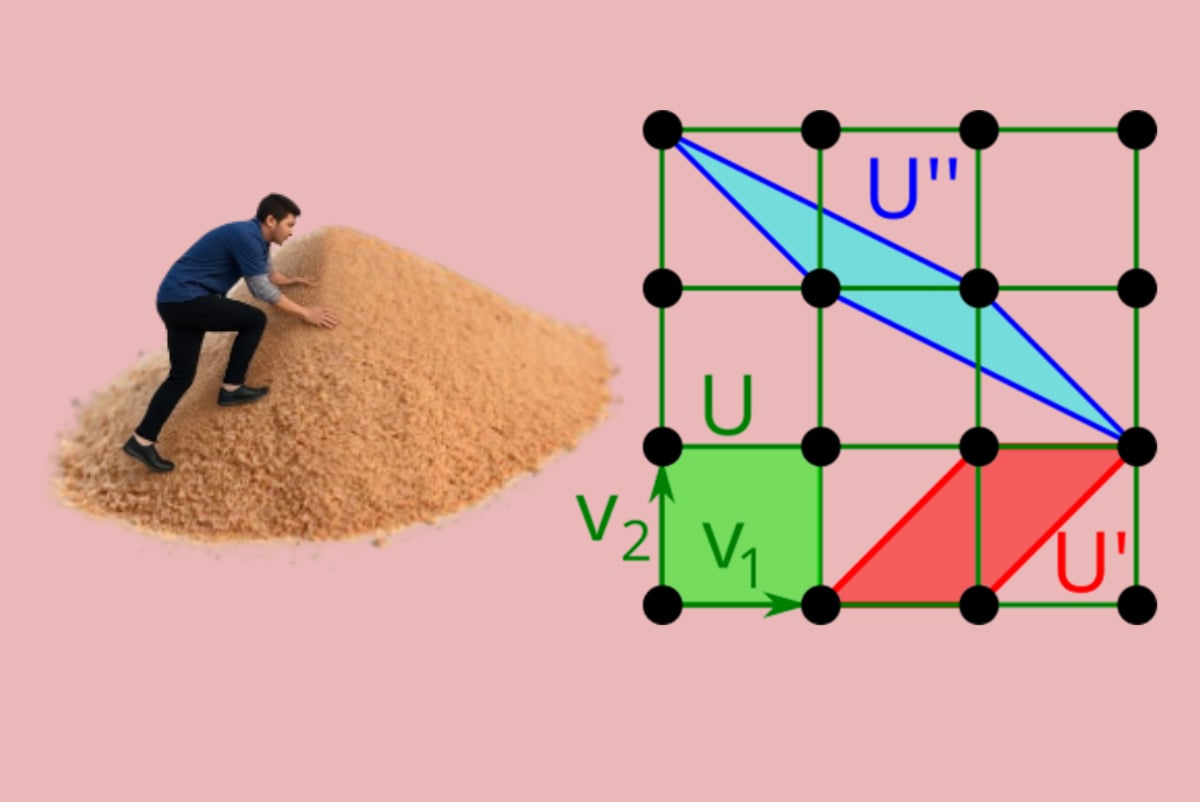
The traditional definition of a periodic crystal starts with a unit cell and a motif of atoms or ions given in fractional coordinates with respect to the unit cell basis, as in a typical Crystallographic Information File (CIF). However, this cell-based representation is highly ambiguous, in that infinitely many pairs (basis, motif) and hence CIFs represent the same periodic crystal (see the illustrative images at the beginning).
But which crystals can (or should) be considered the same?
This question has appeared in the titles of crystallographic papers[4] and can be resolved by explicitly defining an equivalence relation on all periodic crystals as physical objects.
For example, chemists might call crystals (chemically) equivalent if they have the same chemical composition. This makes sense, though we would certainly like to distinguish between diamond and graphite, both of which consist of pure carbon.
Since crystal structures are determined in a rigid form, their natural equivalence is rigid motion (a composition of translations and rotations) that exactly matches all atomic positions. This rigid equivalence is practically the strongest, because there is little sense in distinguishing crystals whose representations differ only by rigid motion.
Motivated by previously incomplete definitions without an explicit equivalence, the paper “The importance of definitions in crystallography”[5] in June 2024 defined a crystal structure as a class of all periodic crystals, i.e. their representations via a basis and a motif as in a CIF, that can be exactly matched with each other by rigid motions.
The crucial difference with past attempts to define isostructural crystals[6] is the new requirement of exact matching justified by the transitivity axiom of an equivalence: if A is equivalent to B, which is equivalent to C, then A is equivalent to C (briefly, if A~B~C, then A~C). Indeed, if atomic perturbations are ignored up to any positive threshold, the transitivity axiom implies that atomic perturbations can be continued and hence a long enough chain of tiny perturbations A_1~A_2~…~A_n can make any objects equivalent.
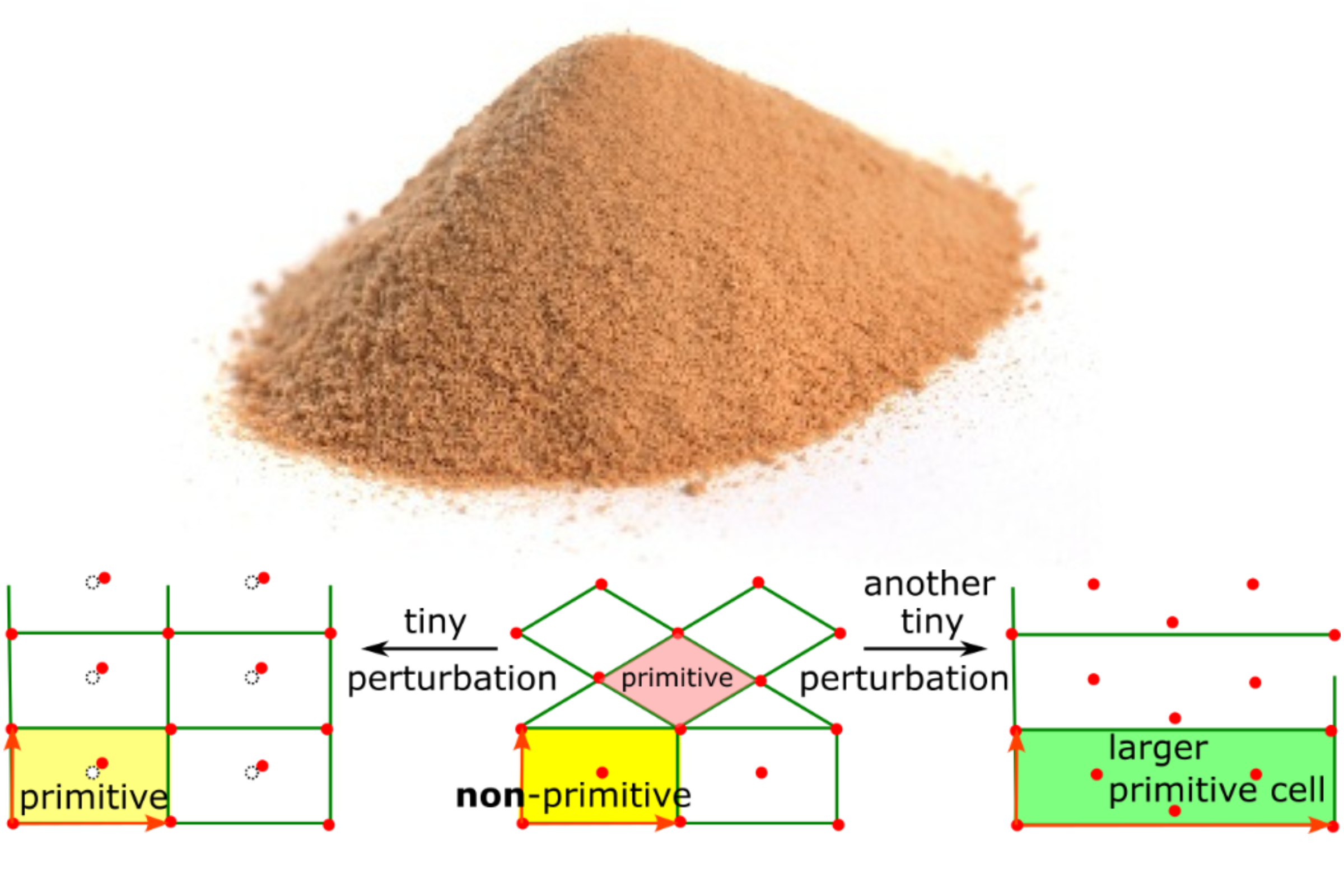
This conclusion has been known since ancient times as the Sorites paradox: when does a heap of (a huge number of grains of) sand stop becoming a heap if grains of sand are removed one by one? See the image below on the left.
The same reasons imply that we should distinguish all periodic crystals that differ by tiny perturbations. In other words, fixing any noise bound and calling crystals pseudo-symmetric only leads to pitfalls [[7]] and shifts the problem from 0 to another threshold.
The image above on the right shows that a primitive cell of any crystal, not only a highly symmetric one, can discontinuously change in size up to an arbitrary integer factor, Hence, any CIF is similar to a 3-dimensional photograph of a real object and should be considered as only one of infinitely many possible representations of the same object.
Like CIFs based on reduced or conventional cells, passport photographs were standardised and used for a long time for identification, but scientific progress has moved us towards more reliable biometric data, while crystallography remained stuck with CIFs.
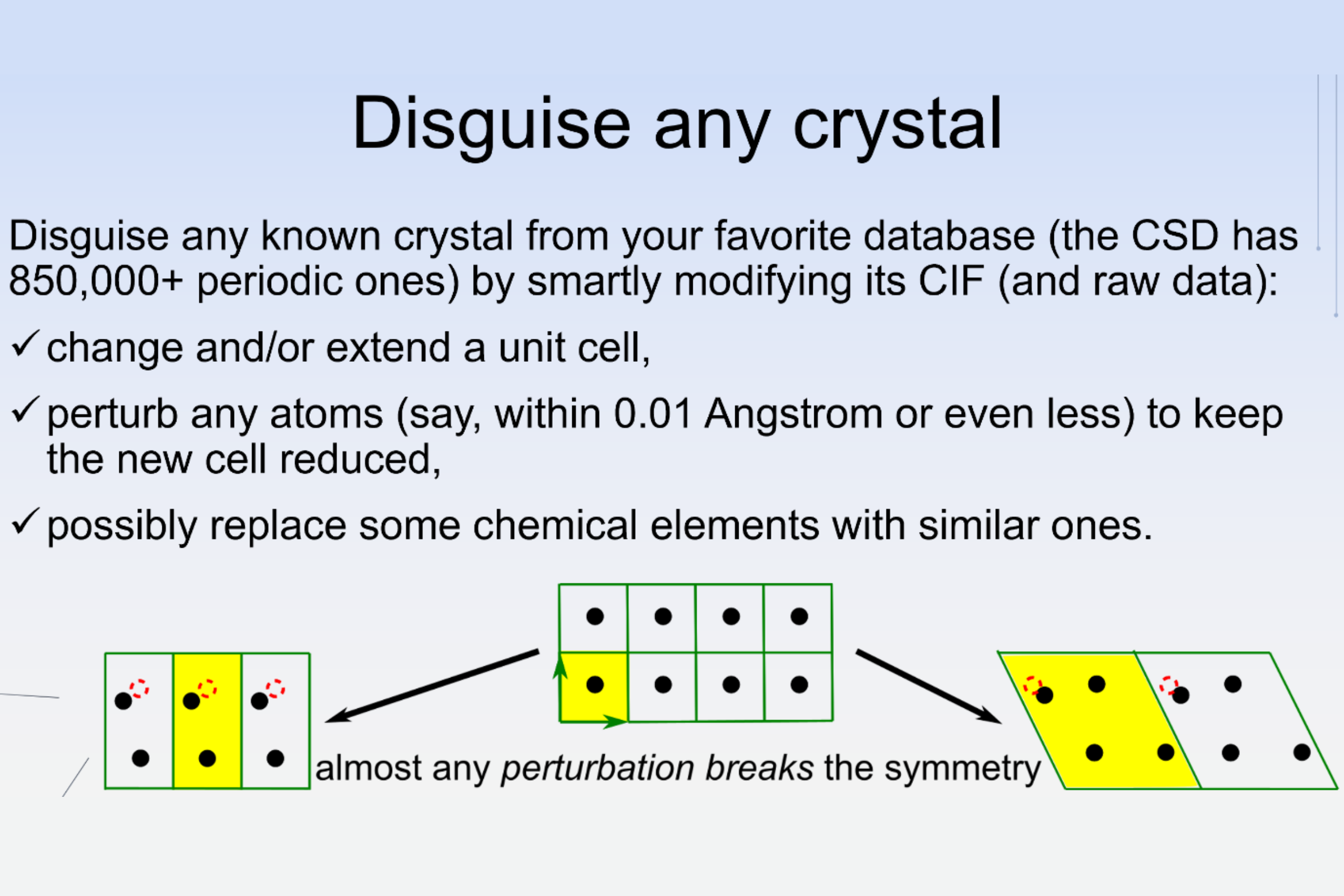
Unlike bounded objects in photographs, noise can substantially change CIFs, and hence any periodic crystal can be easily disguised, as demonstrated in the Science Slam talk “The rise of AI after the fall of AI”, which received the 2nd prize at ECM 2024 in Padova.
The resulting loophole (absence of an equivalence in past definitions of a crystal) led to the duplicate dilemma[2] or the crisis of trust in crystallography[8] when anyone can easily perturb CIFs and structure factors of known crystals and report “a discovery” of a new material, also by changing chemical elements to avoid detection by composition.
While the previous crystal myth[9] led to dozens of retractions in 2010, a few disguises were missed and then exposed in 2021 by mathematicians who first rigorously stated and then practically resolved the data ambiguity problem for periodic crystals[10].
Following the first public version of the foundational paper[10] in September 2020, the IUCr Congress in 2021 included the online talk “A unique and continuous code of all periodic crystals”[11]. See Chapter 12 in the last version of the Geometric Data Science book[12] for more details of mathematical developments in chronological order. In September 2021, the first five duplicate pairs in the CSD were privately reported to the Cambridge Crystallographic Data Centre (CCDC). Although five journals initiated investigations into underlying publications, only two papers have been retracted[13],[14].
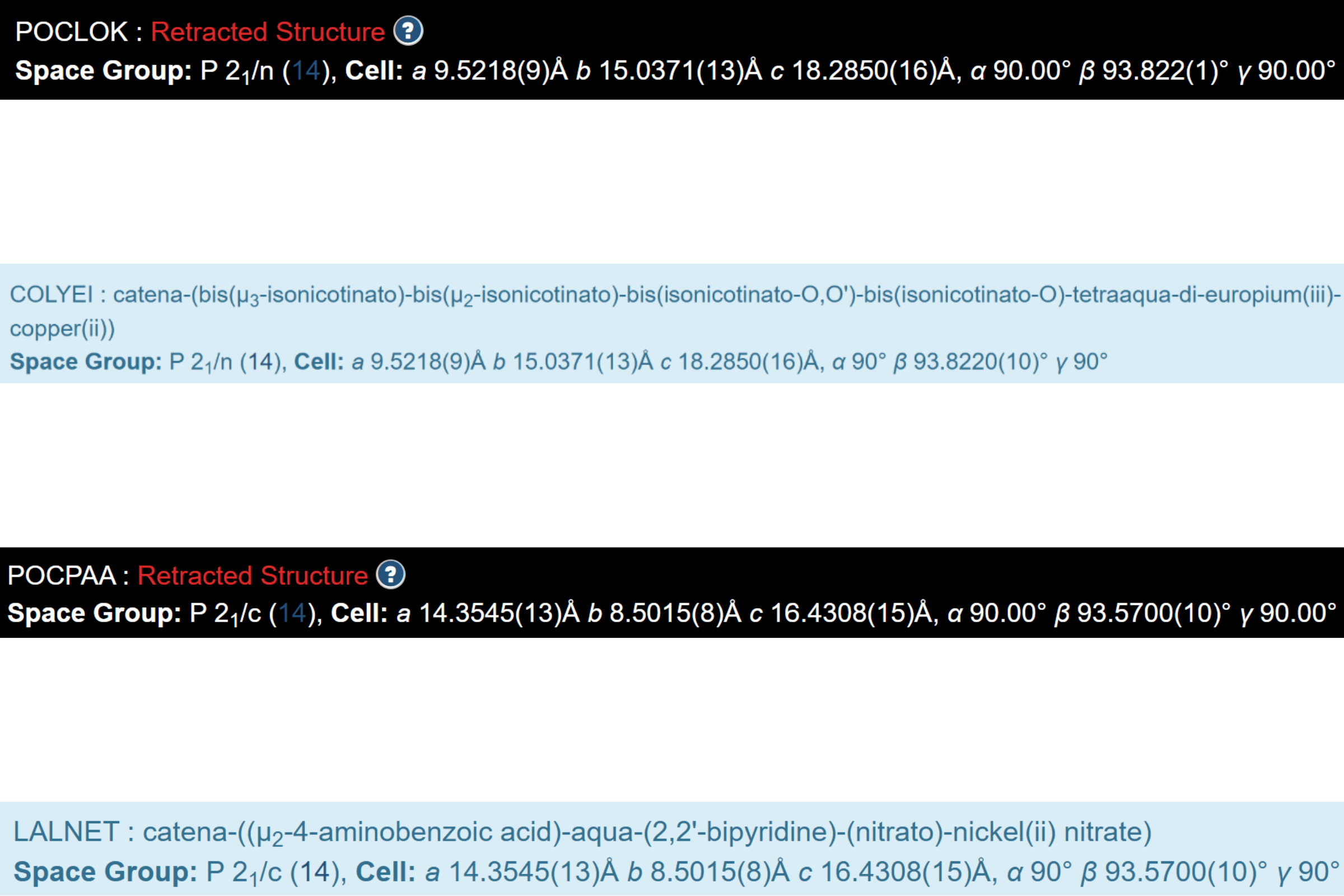
See below the screenshots from the public webpages for two pairs of CSD entries, where one chemical element was replaced with a different one: Sm in POCLOK vs Eu in COLYEI, and Cd in POCPAA vs Ni in LALNET, while keeping other data almost the same.
It remains unclear why POCLOK (not COLYEI) and POCPAA (not LALNET) were retracted, though all numbers in the CIFs of both pairs are identical almost to the last decimal place.
For another pair, DTBIPT vs DTHBPD10, where Pd was replaced with Pt, the original paper[15] in 1976, on page 4 of the supplementary file, honestly acknowledged that the ‘positional parameters for the analogous Pt compound are not significantly different from those above’.
Noise in practical measurements motivates the following answer to the fundamental question, “Same or different?”[4]: real objects should always differ at least slightly, unless their data are copied and pasted, as in the last case above.
For the other two pairs, AFIBOH vs NENCUF and HIFCAB vs JEPLIA, in the initial batch[10] from September 2021, where Cd was replaced with Zn and Mn, respectively, no structures have been retracted because the authors have ignored requests from journals.
The last two pairs were published in 2006-2007 but missed in the 2010 investigation and editorial[16], which started as follows: “Regrettably, this editorial is to alert readers and authors of Acta Crystallographica Section E and the wider scientific community to the fact that we have recently uncovered evidence for an extensive series of scientific frauds involving papers published in the journal, principally during 2007”. In all the cases discussed, the public pages of the CSD entries above still lack links to duplicates and underlying research[10], whereas the retracted papers are well referenced.
Though duplicate structures are now rigorously defined[5] as exactly equivalent under rigid motion, we should talk about near-duplicates in practice because even simple computations of inter-atomic distances accumulate floating-point errors. Hence, any justified comparison of periodic crystals should be provably continuous under perturbations that discontinuously change reduced or conventional cells.
After the previous news article[2], the CCDC updated internal comments for the 5 pairs of near-duplicates discussed above in February 2026, while many more thousands of entries are waiting for investigation, starting with the list that appeared in June 2022 in Table 6 on page 20 of the 2nd version (https://arxiv.org/pdf/2108.04798v2).
While papers[10] and [17] in 2022 practically resolved the data ambiguity for periodic crystals and closed the discontinuity loophole in crystallography by developing generically complete invariants, PDD (Pointwise Distance Distributions), numerous papers continued to use unreliable tools, leading to calls for retraction.
After the CE&N news feature in December 2025[1], Nature published the correction of the so-called A-lab paper[18], where almost all the words “novel” and “discovery” were crossed out.
Leslie Schoop, who, together with Robert Palgrave, has led the first investigation[19] of the original A-lab paper, has started to properly justify the structural novelty of materials[20] by using the local novelty distance in a continuous materials space[21].
All crystallographers are welcome to learn the latest advances[22]-[24] in Geometric Data Science[12] at the 7th MACSMIN (Mathematics and Computer Science for Materials Innovation, ) held on 26-29 May in Liverpool, UK. The underlying area of a continuous crystallography area was first introduced in the talk “The Crystal Isometry Space continuously extends Mendeleev’s table to all periodic materials” at the British Crystallographic Association annual meeting in April 2023.
Since the duplicate dilemma was not answered in the previous newsletter[2], here is the mathematically justified answer to the question “When should experimental crystallographic databases be concerned about near-duplicate crystal structures?”
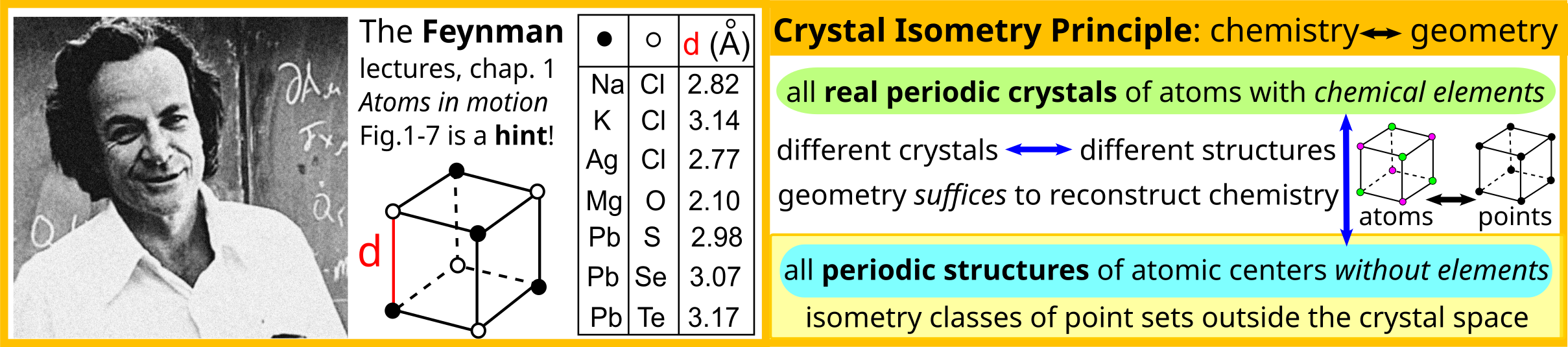
Answer to the “when” question: At least since 1965, because the discontinuity of cell-based representations under perturbations was already appreciated by then. See page 80 in[25], and theoretically, e.g. due to Feynman’s lectures on physics in 1961-1964.
His first lecture, “Atoms in motion”, not only highlighted that atoms vibrate and that their positions are therefore always uncertain, also included Figure 1-7 showing the smallest interatomic distances in 7 cubic crystals. This figure was the crucial hint for stating the Crystal Isometry Principle (CRISP), which holds that precise enough geometry of only atomic centres uniquely determines any real periodic crystal composed of all chemical elements, under fixed ambient conditions, such as temperature and pressure. In other words, chemically different crystals should be distinguishable by geometry alone.
Since May 2021, the CRISP has been experimentally verified on all major materials databases[17], see details in Chapter 12 of the Geometric Data Science book [[12]].
Vitaliy Kurlin, Liverpool University
References
- D.Chawla. Duplicate structures haunt crystallography databases. Chemical & Engineering News, December 2025, https://cen.acs.org/research-integrity/Duplicate-structures-haunt-crystallography-databases/103/web/2025/12. ↩1 ↩2
- N.Johnson et al. Duplicate Dilemma – when should experimental crystallographic databases be concerned about near-duplicate crystal structures? February 2026, https://www.iucr.org/news/newsletter/etc/articles?issue=162129&result_138339_result_page=4. ↩1 ↩2 ↩3 ↩4 ↩5
- C.Brock. Change to the definition of "crystal" in the IUCr Online Dictionary of Crystallography, April 2021, https://www.iucr.org/news/newsletter/etc/articles?issue=151351&result_138339_result_page=17.
- P.Sacchi et al. Same or different–that is the question: identification of crystal forms from crystal structure data." CrystEngComm 22.43 (2020): 7170-7185. ↩1 ↩2
- O.Anosova et al. The importance of definitions in crystallography. IUCrJ, 11(4), 453-463 (2024). ↩1 ↩2
- Isostructural crystals in the IUCr Online Dictionary of Crystallography, https://dictionary.iucr.org/Isostructural_crystals. ↩
- P.Zwart et al. Surprises and pitfalls arising from (pseudo) symmetry." Biological Crystallography 64.1 (2008): 99-107. ↩
- D.Bimler. Better Living through Coordination Chemistry: A descriptive study of a prolific papermill that combines crystallography and medicine, https://doi.org/10.21203/rs.3.rs-1537438/v1 (2022). ↩
- Crystal myth: 11 more retractions from crystallography journal after 2010 fakery, https://retractionwatch.com/2011/02/28/crystal-myth-11-more-retractions-from-crystallography-journal-after-2010-fakery. ↩
- D.Widdowson et al. Average Minimum Distances of periodic point sets are fundamental invariants for mapping all periodic crystals." MATCH Communications in Mathematical and in Computer Chemistry 87(3): 529-559 (2022). ↩1 ↩2 ↩3 ↩4 ↩5
- V.Kurlin. A unique and continuous code of all periodic crystals. Talk at the IUCr congress 2021 (online), 16-min video at https://youtu.be/DNyPSL2JPa4. ↩
- O.Anosova, V.Kurlin. Geometric Data Science (arxiv:2512:05040), the latest version at https://kurlin.org/Geometric-Data-Science-book.pdf. ↩1 ↩2 ↩3
- Retraction of: Yi-Fang Deng, Xue Nie, Chun-Hua Zhang and Dai-Zhi Kuang. Crystal structure of bis[diaquaisonicotinatosamarium(III)]-µ-isonicotinato-[diisonicotinatocopper(II)], CuSm2(C6H4NO2)8(H2O)4. Zeitschrift für Kristallographie – New Crystal Structures, 224: 616–618 (2014), https://doi.org/10.1515/ncrs-2024-0268 ; ↩
- Retraction of: Yi-Fang Deng, Man-Sheng Chen, Chun-Hua Zhang and Dai-Zhi Kuang*. Crystal structure of aqua(2,2′-bipyridine-k 2 N:N′)(nitrato)-(4-aminobenzoato)cadmium(II) nitrate, [Cd(H2O)(NO3)(C10H8N2)(C7H7NO2)][NO3]. Zeitschrift für Kristallographie – New Crystal Structures, 224: 707–708 (2014), https://doi.org/10.1515/ncrs-2024-0269 ; ↩
- R.Girling, E.Amma. Bis (dithiobiureto) MII, M= Pt, Pd. Structural Science 32(10): 2903-2904 (1976). ↩
- W.Harrison et al. Acta Cryst E editorial, 66(1): e1-e2 (2010), https://journals.iucr.org/e/issues/2010/01/00/me0406. ↩
- D.Widdowson, V.Kurlin. Resolving the data ambiguity for periodic crystals. Advances in Neural Information Processing Systems (NeurIPS) 35 (2022): 24625-24638. Extended in SIAP J Appl. Math., to appear at https://doi.org/10.1137/25M1736657, available at https://kurlin.org/projects/periodic-geometry/near-duplicates-materials-databases.pdf. ↩1 ↩2
- Author Correction: An autonomous laboratory for the accelerated synthesis of inorganic materials, Nature 650, E1 (2026), https://static-content.springer.com/esm/art%3A10.1038%2Fs41586-025-09992-y/MediaObjects/41586_2025_9992_MOESM1_ESM.pdf. ↩
- Leeman, Josh, et al. Challenges in high-throughput inorganic materials prediction and autonomous synthesis. PRX Energy 3(1): 011002 (2024). ↩
- X.Zhang et al. Identification of an Unreported Structure Type in GdNiSn4 and Its Implications for Materials Prediction, arXiv:2603.05613 (2026). ↩
- D.Widdowson, V.Kurlin. Geographic-style maps with a local novelty distance help navigate in the materials space. Scientific reports 15(1): 27588 (2025). ↩
- D.Widdowson, V.Kurlin. Continuous invariant-based maps of the Cambridge Structural Database. Crystal Growth & Design, v.24(13), p.5627–5636 (2024). ↩
- O.Anosova et al. Recognition of near-duplicate periodic patterns by continuous metrics with approximation guarantees. Pattern Recognition 171: 112108 (2026).
- S.Majumder et al. Continuous invariant-based asymmetries of periodic crystals quantify deviations from higher symmetry. IUCrJ (2026), to appear, available at https://kurlin.org/projects/periodic-geometry/continuous-asymmetry.pdf. ↩
- S.Lawton, R.Jacobson, R. A. The reduced cell and its crystallographic applications (IS-1141). Ames Lab, Iowa State Univ. of Science and Tech., IA (1965). ↩





![[AsCA logo]](https://www.iucr.org/__data/assets/image/0004/3955/asca.jpg)
![[ECA logo]](https://www.iucr.org/__data/assets/image/0005/3956/ecalogo.gif)