
Acta Cryst. (1996). A52, 770-781![]()
Atomic displacement parameter nomenclature
Report of a Subcommittee on atomic displacement parameter nomenclature
H.-B. BÜRGI, Laboratorium für chemische und mineralogische Kristallographie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland,
H. BURZLAFF, Institut für Kristallographie, Universität, Bismarckstrasse 10, D-91054 Erlangen, Germany,
J.D. DUNITZ, Laboratorium für organische Chemie, ETH Zentrum, CH-8092 Zürich, Switzerland,
C.M. GRAMACCIOLI, Dipartimento di Scienze della Terra, via Botticelli 23, I-20133 Milano,Italy,
H.H. SCHULZ, Institut für Kristallographie und Mineralogie, Universität, Theresienstrasse 41, D-80333 Mü, Germany,
U. SHMUELI, School of Chemistry, Tel Aviv University, 69 978 Tel Aviv, Israel, and
S.C. ABRAHAMS (ex officio, IUCr Commission on Crystallographic Nomenclature), Physics Department, Southern Oregon State College, Ashland, OR 97520, USA.
Abstract
Modern x-ray and neutron diffraction techniques can give precise parameters that describe dynamic or static displacements of atoms in crystals. However, confusing and inconsistent terms and symbols for these quantities occur in the crystallographic literature. This report discusses various forms of these quantities, derived from probability density functions and based on Bragg diffraction data, both when the Gaussian approximation is appropriate and when it is not. The focus is especially on individual atomic anisotropic displacement parameters (ADPs), which may represent atomic motion and possible static displacive disorder.
The first of the four sections gives background information, including definitions. The second concerns the kinds of parameters describing atomic displacements that have most often been used in crystal structure analysis and hence are most commonly found in the literature on the subject. It includes a discussion of graphical representations of the Gaussian mean-square displacement matrix. The third section considers the expressions used when the Gaussian approximation is not adequate. The final section gives recommendations for symbols and nomenclature.
![]()
Established 15 February 1994 by the IUCr Commission on Crystallographic Nomenclature, with all members appointed by 7 June 1994, under the ground rules outlined in Acta Cryst. (1979). A35, 1072. H. Burzlaff resigned from the Subcommittee 2 January 1995. Final Report accepted 20 December 1995 by the IUCr Commission on Crystallographic Nomenclature.
We are especially indebted to Verner Schomaker for his critical reading and counsel on many drafts of this Report, to Hans Boysen for his special help with Section 3, and to Richard Marsh, Emily Maverick and Tullio Pilati for their advice. ![]()
1. Introduction
1.1 Organization of this Report
There are four main sections. The Introduction continues, after the present subsection, with a discussion of the rationale for and genesis of the Subcommittee that wrote this report (Subsection 1.2), notation for position and displacement vectors (1.3), a consideration of the structure factor and atomic displacements (1.4), and comments about the terminology to be used (1.5).
The second main section, Displacement Parameters Based on the Gaussian Approximation, concerns the kinds of parameters describing atomic displacements that have most often been used in crystal structure analysis and hence are most commonly found in the literature on the subject. Its subsections are: 2.1, Anisotropic displacement parameters, which includes a discussion of the common symbols used; 2.2, Equivalent isotropic displacement parameters; and 2.3, Graphical representations of the Gaussian mean-square displacement matrix.
The third main section, Beyond the Gaussian Approximation, considers the expressions used when the Gaussian approximation is not adequate. Our Recommendations for nomenclature are in the final main section.
1.2 Background
Anisotropic displacement parameters (ADPs) (see the discussion of terminology in subsection 1.5 and of symbols in subsection 2.1) have typically been determined and reported in the crystallographic literature in a variety of ways, symbolized sometimes as U (as in ![]() or
or ![]() ), sometimes as B (as in
), sometimes as B (as in ![]() or
or ![]() ), and sometimes as
), and sometimes as ![]() (as in
(as in ![]() ). Unfortunately, these terms have not always been used consistently. Dimensionless quantities, e.g., the
). Unfortunately, these terms have not always been used consistently. Dimensionless quantities, e.g., the ![]() , have been misidentified as
, have been misidentified as ![]() values, which are of dimension (length)2. When these and related quantities were first determined more than four decades ago, they were considered at best to have qualitative significance, and inconsistencies, when noticed, were often disregarded. Cruickshank's (1956) papers marked a turning point toward quantitative interpretation in terms of simple physical models. Since then, technical improvements, both in the measurement of diffraction data and in computing, have led to increased physical significance in the experimentally determined ADPs. The fact that these quantities are now more meaningful, and the lack of uniformity with which they have been presented, have been discussed informally in recent years by more than one IUCr Commission, but no consensus has previously been reached on an acceptable form of presentation. This situation was brought recently to the attention of the Commission on Crystallographic Nomenclature. That body established this Subcommittee, charging it with examining the merits of adopting a uniform approach to reporting in structural papers quantities that describe atomic displacement, and with making such recommendations as might be deemed appropriate. Discussion within the Subcommittee, conducted principally through electronic correspondence, identified two major areas
values, which are of dimension (length)2. When these and related quantities were first determined more than four decades ago, they were considered at best to have qualitative significance, and inconsistencies, when noticed, were often disregarded. Cruickshank's (1956) papers marked a turning point toward quantitative interpretation in terms of simple physical models. Since then, technical improvements, both in the measurement of diffraction data and in computing, have led to increased physical significance in the experimentally determined ADPs. The fact that these quantities are now more meaningful, and the lack of uniformity with which they have been presented, have been discussed informally in recent years by more than one IUCr Commission, but no consensus has previously been reached on an acceptable form of presentation. This situation was brought recently to the attention of the Commission on Crystallographic Nomenclature. That body established this Subcommittee, charging it with examining the merits of adopting a uniform approach to reporting in structural papers quantities that describe atomic displacement, and with making such recommendations as might be deemed appropriate. Discussion within the Subcommittee, conducted principally through electronic correspondence, identified two major areas ![]() in which uniform definitions, terminology, and nomenclature would be desirable. These include anisotropic displacement parameters in the Gaussian approximation and in situations in which the Gaussian approximation is inadequate. In considering the uncertainties of experimentally determined ADPs, the reader is referred to the Report of a Working Group on the Expression of Uncertainty in Measurement (Schwarzenbach, et al., 1995), appointed by the IUCr Commission on Crystallographic Nomenclature, which discusses the general concept of uncertainty in the result of a crystallographic measurement, and its application.
in which uniform definitions, terminology, and nomenclature would be desirable. These include anisotropic displacement parameters in the Gaussian approximation and in situations in which the Gaussian approximation is inadequate. In considering the uncertainties of experimentally determined ADPs, the reader is referred to the Report of a Working Group on the Expression of Uncertainty in Measurement (Schwarzenbach, et al., 1995), appointed by the IUCr Commission on Crystallographic Nomenclature, which discusses the general concept of uncertainty in the result of a crystallographic measurement, and its application.
The recommendations at the end of this report are proposed for use in all future publications of the International Union of Crystallography. Authors of structure refinement programs, particularly those in widespread use, are encouraged to bring their programs into full conformity with these recommendations.
![]()
Recommendations on the nomenclature of thermal diffuse scattering [e.g., Willis (1993)] and disorder diffuse scattering [e.g., Jagodzinski & Frey (1993)] were considered as outside the scope of this report.
1.3 Notation for position and displacement vectors
Some notation to be used in this report for basis vectors and their components is summarized here. The local instantaneous atomic position vector is denoted by r+u, with r the mean atomic position vector and u the displacement vector of an atom from its mean position. We use the symbols ![]() , and
, and ![]() , as is common, for the lengths of the reciprocal axes.
, as is common, for the lengths of the reciprocal axes.
Quantities referred to the direct lattice basis, a, b, c (or equivalently ![]() ) :
) :
Components of r: x, y, z (or equivalently ![]() ).
).
Components of u: ![]() (or equivalently
(or equivalently ![]() ).
).
Related anisotropic displacement parameter: ![]() ; (dimensionless).
; (dimensionless).
Quantities referred to the basis ![]() (or equivalently
(or equivalently ![]() ):
):
Components of r: ![]() (or equivalently
(or equivalently ![]() ).
).
Components of u: ![]() (or equivalently
(or equivalently ![]() ).
).
Related anisotropic displacement parameter: ![]() ; (length)2.
; (length)2.
Quantities referred to the Cartesian basis ![]() ):
):
Components of r: ![]() (or equivalently
(or equivalently ![]() ).
).
Components of u: ![]() (or equivalently
(or equivalently ![]() ).
).
Related anisotropic displacement parameter: ![]() ; (length)2.
; (length)2.
1.4 The structure factor and atomic displacements
The structure factor of reflection h is given in a fairly general form by the Fourier transform of the average density of scattering matter
with the integration extending over the repeating structural motif, confined to a single unit cell. The brackets denote a double averaging over the possible displacements of the atoms from their mean positions - a time average over the atomic vibrations in each cell, followed by a space average that consists of projecting all the time-averaged cells onto one and dividing by the number of cells, h is a diffraction vector obeying the Laue equations, and ![]() is the static density of the motif, consistent with the instantaneous local configuration of the nuclei in a unit cell.
is the static density of the motif, consistent with the instantaneous local configuration of the nuclei in a unit cell.
To reduce the above general picture to what is used in conventional crystal structure analysis, we first assume that the average density of matter in (1.4.1) can be regarded as a superposition of averaged atomic densities. This so-called isolated-atom approximation is essentially equivalent to assuming independently displaced atoms, a fair initial approximation, although not generally valid. The average density of scattering matter at the point r in a unit cell can then be approximated as
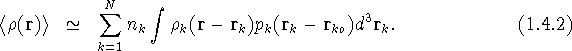
Here N is the number of atoms in the unit cell, ![]() is the occupancy factor of the kth atom,
is the occupancy factor of the kth atom, ![]() is the density (electron density for X-rays, or a delta function weighted with the scattering length
is the density (electron density for X-rays, or a delta function weighted with the scattering length ![]() for neutrons) due to atom k at a point r when the nucleus of atom k is at
for neutrons) due to atom k at a point r when the nucleus of atom k is at ![]() , and
, and ![]() is the probability density function (pdf) corresponding to the probability for having atom k displaced by the vector (
is the probability density function (pdf) corresponding to the probability for having atom k displaced by the vector ( ![]() -
- ![]() ) from its reference position
) from its reference position ![]() in an average unit cell, which will be the mean position if
in an average unit cell, which will be the mean position if ![]() is sufficiently symmetrical. It is important to remember that the approximations in (1.4.2) include the assumption that atoms are not deformable, by bonding or otherwise, even though at this stage the static atomic electron density,
is sufficiently symmetrical. It is important to remember that the approximations in (1.4.2) include the assumption that atoms are not deformable, by bonding or otherwise, even though at this stage the static atomic electron density, ![]() , has not been assumed to be spherically symmetrical.
, has not been assumed to be spherically symmetrical.
If eq. (1.4.2) is now substituted into eq. (1.4.1), and the order of the summation and integration is interchanged, the structure factor becomes

with
If the substitutions ![]() and
and ![]() are made, the integral in (1.4.4) becomes
are made, the integral in (1.4.4) becomes
The inner integral in (1.4.5) has the form of a conventional convolution of the density of atom k with the pdf for a displacement of this atom from its mean position; the outer integral is a Fourier transform of this convolution. This transform is multiplied by an exponential that depends on the mean position, ![]() , of atom k.
, of atom k.
By the convolution theorem, the Fourier transform of a convolution equals the product of Fourier transforms of the functions involved. When this theorem is applied to the outer integral in (1.4.5), we obtain the conventional approximation for the structure factor of a Bragg reflection
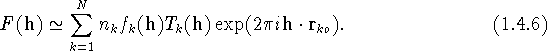
If we let ![]() and (as before)
and (as before) ![]() , then in (1.4.6)
, then in (1.4.6)
is the scattering factor or form factor of atom k (for neutrons this is replaced by the scattering length ![]() ), and
), and
is the Fourier transform of the pdf, ![]() , for the displacement of the kth atom from its reference position, r
, for the displacement of the kth atom from its reference position, r ![]() . This term contains the dependence of the structure factor on atomic displacements, and has been known by the names ``atomic Debye-Waller factor" and ``atomic temperature factor" (see section 1.5). There are no restrictions on the functional form of the pdf in the integrand of (1.4.8).
. This term contains the dependence of the structure factor on atomic displacements, and has been known by the names ``atomic Debye-Waller factor" and ``atomic temperature factor" (see section 1.5). There are no restrictions on the functional form of the pdf in the integrand of (1.4.8).
Let us now recall that the structure factor equation used in routine refinement of atomic parameters is further simplified in two ways:
First, for X-rays, the static atomic electron density is assumed to have spherical symmetry. This reduces the atomic scattering factor to the form
which has been computed and extensively tabulated for all the neutral elements and many ions (Maslen, Fox & O'Keefe, 1992). The spherical-atom approximation necessarily removes fine details of the (calculated) electron density, but may be used routinely, and serve as a starting point for more refined determinations of atomic positions and studies of charge density (e.g., Coppens & Becker, 1992; Coppens, 1993).
Second, the pdf for atomic displacement is most frequently approximated by a univariate or trivariate Gaussian, depending on whether the atomic displacements are assumed to be isotropic or anisotropic respectively. If a trivariate Gaussian is assumed, and the atomic subscript k is omitted, the resulting expression for ![]() from (1.4.8) is
from (1.4.8) is
Equation (1.4.10) can be derived from the theory of lattice dynamics in the harmonic approximation, which considers only the (always present) contribution of motion to the atomic displacement (e.g., Willis & Pryor, 1975). However, this equation may also be applied to static displacive disorder. The form of the atomic Debye-Waller factor, ![]() , represented in (1.4.10) is the most common one in standard structure refinements and will be discussed in Section 2. Various other approximations have been proposed for situations in which the Gaussian formalism is not adequate, e.g., when the anharmonic contribution to the crystal dynamics is significant; the most common are discussed in section 3.
, represented in (1.4.10) is the most common one in standard structure refinements and will be discussed in Section 2. Various other approximations have been proposed for situations in which the Gaussian formalism is not adequate, e.g., when the anharmonic contribution to the crystal dynamics is significant; the most common are discussed in section 3.
We present now a short discussion of common variants of eq. (1.4.10), which can be rewritten as
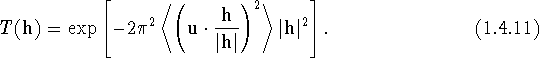
This shows that the exponent is proportional to minus the mean-square projection of the atomic displacement u on the direction of the diffraction vector h times the squared magnitude of h. If we denote the projection of u on the direction of h by ![]() , and make use of the relation:
, and make use of the relation: ![]() , (1.4.11) becomes
, (1.4.11) becomes
As long as the atomic displacements are anisotropic, the value of the average in (1.4.12) depends on the direction of h. This is then the anisotropic Gaussian Debye-Waller factor, ![]() , which is discussed in detail in Section 2. If, however, the atomic displacements are isotropic, the average in (1.4.12) is a constant determined by the structure alone, but possibly different for non-equivalent atoms, and the left-hand side of this equation no longer depends on the direction of h, but only on its magnitude. This is then the atomic isotropic Gaussian Debye-Waller factor,
, which is discussed in detail in Section 2. If, however, the atomic displacements are isotropic, the average in (1.4.12) is a constant determined by the structure alone, but possibly different for non-equivalent atoms, and the left-hand side of this equation no longer depends on the direction of h, but only on its magnitude. This is then the atomic isotropic Gaussian Debye-Waller factor,
The lowest-order approximation to ![]() is the overall isotropic Debye-Waller factor. It has the same form as (1.4.13), and presumes that all the atoms have the same isotropic mean-square displacement,
is the overall isotropic Debye-Waller factor. It has the same form as (1.4.13), and presumes that all the atoms have the same isotropic mean-square displacement, ![]() . The whole crystal structure is assigned, in this approximation, a single displacement parameter. This approximation is used in initial stages of crystal structure determination by direct methods.
. The whole crystal structure is assigned, in this approximation, a single displacement parameter. This approximation is used in initial stages of crystal structure determination by direct methods.
We conclude this section with some remarks on the structure factor for electron diffraction by a crystal. The density of scattering matter, ![]() , is here interpreted as the distribution of electrostatic potential within the unit cell. This potential is then approximated by a superposition of electrostatic potentials contributed by individual atoms, and the effects of motion are taken into account, as for X-rays and neutrons, by the convolution of the potential of an atom at rest with the probability density function describing the atomic motion (e.g., Vainshtein and Zvyagin, 1993). The atomic (spherical) scattering factor for electron diffraction,
, is here interpreted as the distribution of electrostatic potential within the unit cell. This potential is then approximated by a superposition of electrostatic potentials contributed by individual atoms, and the effects of motion are taken into account, as for X-rays and neutrons, by the convolution of the potential of an atom at rest with the probability density function describing the atomic motion (e.g., Vainshtein and Zvyagin, 1993). The atomic (spherical) scattering factor for electron diffraction, ![]() , for an atom at rest and diffraction vector
, for an atom at rest and diffraction vector ![]() , is related to that for X-rays by the Mott formula (e.g., Vainshtein, 1964) which has the form
, is related to that for X-rays by the Mott formula (e.g., Vainshtein, 1964) which has the form
![]()
with ![]() the atomic number and
the atomic number and ![]() is the X-ray form factor of atom k [see (1.4.9)]. This formula, with the correct proportionality constants, has been used along with other techniques in extensive tabulations of spherical form factors for electron-diffraction (see, e.g., Cowley, 1992). The Debye-Waller factor, here expressing the `smearing out' of the electrostatic potential, is given by the same expression as that quoted above for X-rays and neutrons (e.g., Vainshtein, 1964; Vainshtein & Zvyagin, 1993). The structure factor for electron diffraction is therefore analogous to that appearing in (1.4.6) but is often given in a different notation.
is the X-ray form factor of atom k [see (1.4.9)]. This formula, with the correct proportionality constants, has been used along with other techniques in extensive tabulations of spherical form factors for electron-diffraction (see, e.g., Cowley, 1992). The Debye-Waller factor, here expressing the `smearing out' of the electrostatic potential, is given by the same expression as that quoted above for X-rays and neutrons (e.g., Vainshtein, 1964; Vainshtein & Zvyagin, 1993). The structure factor for electron diffraction is therefore analogous to that appearing in (1.4.6) but is often given in a different notation.
1.5 Comments about terminology
The quantity T that occurs in eq. (1.4.6) has been commonly referred to either as the Debye-Waller factor or the temperature factor because Debye (1913) and Waller (1923) first understood and formulated the effect that thermal vibrations would have on the intensity of X-ray scattering. It has, however, long been recognized, as discussed in section 1.4 above, that static displacements would have a similar effect. We therefore avoid the term ``temperature factor", and recommend that others do so also, in part because of this ambiguity about the origin of the atomic displacements that cause the diminution in scattering. Another reason for avoiding the phrase ``temperature factor" is the confusion caused by the fact that it has not infrequently in the past been used for terms in the exponent in expressions like that on the right side of eqs. (1.4.12) and (1.4.13), rather than for the entire exponential multiplicative factor.
A detailed treatment of the physical background of possible atomic displacements is quite beyond the scope of this report. However, we shall try to summarize and describe briefly the most important components of the displacement. The best known is the displacement arising from atomic vibrations. When these result from the motion of molecules or molecular fragments (e.g., Willis & Pryor, 1975), they are usually characterized by relatively large amplitudes. In crystals containing relatively strongly bonded atoms (e.g., molecular and ionic crystals), much smaller displacement amplitudes result from the ever-present internal vibrations, such as bond stretching and bending (e.g., Wilson, Decius, & Cross, 1954). All of these motions are temperature-dependent, unless the temperature is very low. Other effective displacements from the mean position may arise as a result of a variety of possible types of disorder. These include small deviations from ideal periodicity, present in all real crystals; orientational disorder, present in many molecular crystals; density and displacement modulations; and short- and long-range displacive correlations. Many types of disorder give rise to diffuse scattering, which can often be analyzed (e.g., Jagodzinski & Frey, 1993). There are, in addition, numerous other possible contributions to apparent displacements, one of the most important of which is use of an inadequate model, e.g., inadequate absorption correction, or use of a Gaussian probability density function when it is inappropriate.
In view of the large number of possible causes of an apparent atomic displacement, we recommend expanding the definition of ``Debye-Waller factor" to include displacements arising from any source. We will use the term ``Debye-Waller factor" when we mean the entire factor that multiplies the scattering factor of an atom at rest, and recommend that this term be used when words are wanted to refer to the quantity ![]() , or T, that occurs in equations such as (1.4.6), (1.4.8), and (1.4.10) through (1.4.13).
, or T, that occurs in equations such as (1.4.6), (1.4.8), and (1.4.10) through (1.4.13).
There was considerable discussion in our Subcommittee concerning the proper words to use when referring to the terms in the exponent that are variables during a typical least-squares refinement to fit a structural model to intensity data. These terms are formulated and symbolized in various ways, discussed in detail in section 2.1 below. We recommend unanimously the term ``displacement parameters" (often ``anisotropic displacement parameters", or ADPs) to describe these quantities. Two of us initially favored ``displacement coefficients", believing that once refinement is completed, this term is more appropriate, but were persuaded that current usage strongly favors the recommended term, ``displacement parameters".
2. Displacement Parameters Based on the Gaussian Approximation
2.1 Anisotropic displacement parameters
As discussed in section 1.4 above, diffraction studies yield information not only about mean atomic positions but also about the probability density functions (pdf's) of atomic displacements from these mean positions. If the atomic pdf is assumed to be a trivariate Gaussian, the characteristic function corresponding to this pdf - by definition, its Fourier transform - can be described by the second moments of the pdf, which in the present context are called anisotropic mean-square displacements. If not, higher cumulants of a non-Gaussian pdf can, in principle, also be determined; these are simple functions of moments (e.g., Kendall & Stuart, 1977), but there are difficulties. For example, these higher terms are only likely to be important when the second moments of the pdf's are relatively large. However, as can be seen from the basic expression for the isotropic Debye-Waller factor, ![]() , the larger the second moment, the more rapidly the scattering from the atomic center in question falls off with increase in the scattering angle. Thus, just when the higher terms become important, they become difficult to measure, for lack of contribution by the scattering center to the Bragg intensities.
, the larger the second moment, the more rapidly the scattering from the atomic center in question falls off with increase in the scattering angle. Thus, just when the higher terms become important, they become difficult to measure, for lack of contribution by the scattering center to the Bragg intensities.
The mean-square displacements, which define the pdf in the various Gaussian approximations, used to be known as atomic vibration parameters or thermal parameters but have recently been designated as atomic displacement parameters, isotropic or anisotropic, to allow for the effects of static displacive disorder, as well as for those of the always-present atomic motion. There exists an extensive literature on the interpretation of these parameters (e.g., Dunitz, Schomaker & Trueblood, 1988, and references cited therein).
The purpose of this section is to relate alternative forms of anisotropic displacement parameters (ADPs) to the expression for the Debye-Waller factor that is valid within the framework of the assumptions underlying the harmonic approximation (e.g., Willis & Pryor, 1975). We also discuss anisotropic displacement parameters in relation to different coordinate systems, outline the transformation properties of the resulting quantities, present several forms of equivalent isotropic displacement parameters, and describe briefly graphical representations of the Gaussian mean-square displacement matrix.
The usual expression for T(h) is [eq. (1.4.10) restated]:
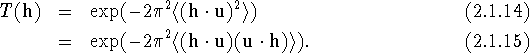
These fundamental equations take on different forms according to the basis vectors to which we refer the diffraction and displacement vectors. In carrying out coordinate transformations in the formalism of tensor algebra, quantities that transform like direct basis vectors are called covariant and are indicated by subscripts, while quantities transforming like reciprocal basis vectors are called contravariant and are indicated by superscripts. The direct and reciprocal bases are not necessarily those of the corresponding lattices; they may be any pair of dual bases. Let us first assume that the diffraction vector is referred to the basis of the reciprocal lattice and the atomic displacement vector to the basis of the direct lattice, as follows
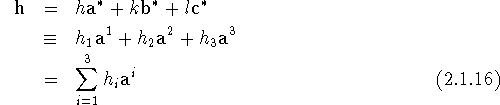
and
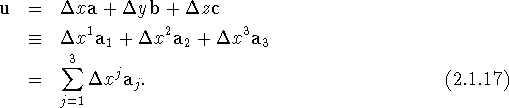
Note that the components of h and u are dimensionless. The first scalar product appearing on the right-hand side of eq. (2.1.15) can now be evaluated as
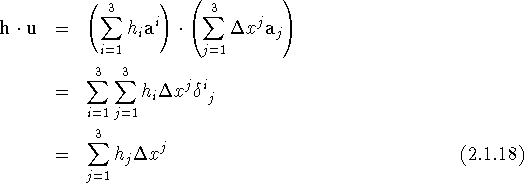
and similarly

We used here the definition of the dual (direct and reciprocal) bases:
![]()
If we insert eqs. (2.1.18) and (2.1.19) into eq. (2.1.15) we obtain for ![]()
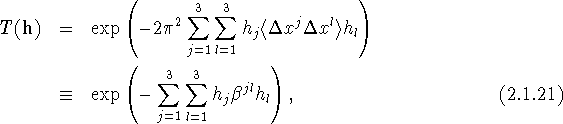
with
The quantity ![]() defined by eqs. (2.1.21) and (2.1.22) is one of the frequently employed forms of the anisotropic displacement parameter; note the use of superscripts for the indices, since the components of
defined by eqs. (2.1.21) and (2.1.22) is one of the frequently employed forms of the anisotropic displacement parameter; note the use of superscripts for the indices, since the components of ![]() are contravariant. For an atom, each component
are contravariant. For an atom, each component ![]() is 2
is 2 ![]() times an average of a product of two components of an atomic displacement vector, when the latter is referred to the basis of the direct lattice.
times an average of a product of two components of an atomic displacement vector, when the latter is referred to the basis of the direct lattice.
We shall now retain h as defined by eq. (2.1.16), but redefine u as follows
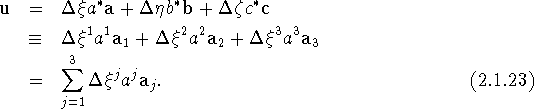
The components of u in this representation, ![]() , have dimension length, and the basis vectors
, have dimension length, and the basis vectors ![]() are dimensionless (see, e.g., Hirshfeld & Rabinovich, 1966). Only in orthorhombic, tetragonal and cubic crystal systems must these basis vectors be mutually orthogonal unit vectors, i.e., orthonormal, since it is only in these systems that the equalities
are dimensionless (see, e.g., Hirshfeld & Rabinovich, 1966). Only in orthorhombic, tetragonal and cubic crystal systems must these basis vectors be mutually orthogonal unit vectors, i.e., orthonormal, since it is only in these systems that the equalities ![]() ,
, ![]() , and
, and ![]() are necessarily true. The departures of these basis vectors from orthonormality in other systems are associated with the departures of the angles
are necessarily true. The departures of these basis vectors from orthonormality in other systems are associated with the departures of the angles ![]() ,
, ![]() , and
, and ![]() from 90
from 90 ![]() . If we now repeat the evaluation of the scalar products in eq. (2.1.15) with h given by eq. (2.1.16) and u given by eq. (2.1.23), we obtain for T
. If we now repeat the evaluation of the scalar products in eq. (2.1.15) with h given by eq. (2.1.16) and u given by eq. (2.1.23), we obtain for T
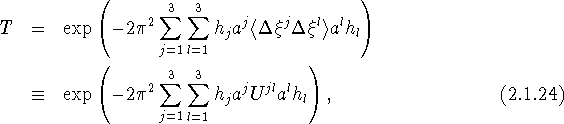
with
another well known form of the ADP. This form is often preferred because the elements of the tensor ![]() U have dimension (length)2 and can be directly associated with the mean-square displacements of the atom considered in the corresponding directions. Note in particular that the mean-square displacement in an arbitrary direction denoted by the unit vector n, when n is referred to unit vectors parallel to the reciprocal basis vectors so that its components are covariant, is given by
U have dimension (length)2 and can be directly associated with the mean-square displacements of the atom considered in the corresponding directions. Note in particular that the mean-square displacement in an arbitrary direction denoted by the unit vector n, when n is referred to unit vectors parallel to the reciprocal basis vectors so that its components are covariant, is given by ![]() (see section 2.3.2). In any event, the dimensionless elements of
(see section 2.3.2). In any event, the dimensionless elements of ![]() are also correctly associated with the general expression for T(h), given by eq. (2.1.15).
are also correctly associated with the general expression for T(h), given by eq. (2.1.15).
Another form of the anisotropic displacement parameter, which is used in some conventional refinement calculations, especially in biomolecular crystallography, is
and the corresponding expression for T becomes
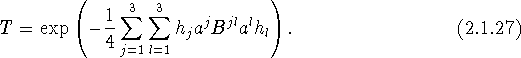
Since B and U are equivalent, apart from a constant factor, and U has a more direct physical significance than B, we recommend that the use of B be discouraged.
A brief discussion of the transformation properties of ![]() and U may be helpful. The corresponding representations of the atomic displacement vector are
and U may be helpful. The corresponding representations of the atomic displacement vector are

and

respectively (Hirshfeld & Rabinovich, 1966). If the basis of the direct lattice is changed in some manner, the new components of the displacement vector u are related to the old ones by linear transformations, say

and

The elements of the transformation matrices depend on the old and new bases. It follows from eqs. (2.1.22) and (2.1.25) that ![]() and U transform as products of the corresponding components of the displacement vector. Hence, the transformation rules for
and U transform as products of the corresponding components of the displacement vector. Hence, the transformation rules for ![]() and U become
and U become

and

and thus conform to those valid for tensors of the second order (e.g., Spain, 1956). The transformation matrices R and Q are obviously different, since the basis vectors to which u, in its two representations, is referred depend in a different manner on the basis of the direct lattice . This transformation property will be illustrated in detail in section 2.1.2 by the orthogonalization of ![]() and U.
and U.
We comment finally on the form of the Debye-Waller factor when both the diffraction vector and the atomic displacement vector are referred to the same Cartesian basis, say ![]() ,
, ![]() ,
, ![]() . It is understood that the use of this representation is usually, in crystallographic practice, preceded by appropriate transformations (see below for a detailed example).
. It is understood that the use of this representation is usually, in crystallographic practice, preceded by appropriate transformations (see below for a detailed example).
The h and u vectors, in the Cartesian representation, are given by
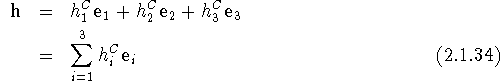
and
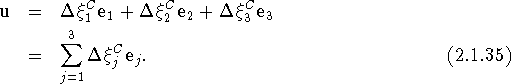
All the indices are given here as subscripts, since in the Cartesian representation the position of the indices is irrelevant. Note that the components of h in eq. (2.1.34) have dimension (length)2. The scalar products in eq. (2.1.15) are now readily evaluated and we obtain for the Debye-Waller factor
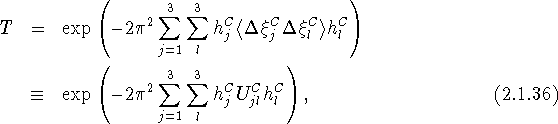
with
an element of an atomic mean-square displacement tensor, with dimension (length)2, referred to a Cartesian basis. This representation avoids the hazards associated with calculations in oblique coordinate systems and is used almost always in lattice-dynamical studies and thermal motion analysis, and very often in constrained refinement of atomic parameters.
![]()
To be precise, the symbols U and
2.1.1 Relationships between the anisotropic displacement parameters
The displacement parameters ![]() ,
, ![]() , and
, and ![]() , given by eqs. (2.1.22), (2.1.25), and (2.1.26), are so closely related that the use of all of them in the crystallographic literature is not only unnecessary, but conducive to confusion. The relationships needed are shown in a single equation, obtained by comparison of eqs. (2.1.21), (2.1.24) and (2.1.27):
, given by eqs. (2.1.22), (2.1.25), and (2.1.26), are so closely related that the use of all of them in the crystallographic literature is not only unnecessary, but conducive to confusion. The relationships needed are shown in a single equation, obtained by comparison of eqs. (2.1.21), (2.1.24) and (2.1.27):
It is true that fewer arithmetic operations are needed to compute T from ![]() than from
than from ![]() , but with the advent of computers this advantage has become negligible.
, but with the advent of computers this advantage has become negligible.
2.1.2 Construction of Cartesian mean-square displacement tensors
Referring an ADP tensor to a Cartesian basis is somewhat less simple. We proceed to show how this is done, both in order to illustrate the above-outlined transformation of tensors and to provide some background for the following section.
Construct a Cartesian system by taking, for example, ![]() along a,
along a, ![]() along
along ![]() , and
, and ![]() along the vector product
along the vector product ![]() . The resulting vectors
. The resulting vectors

comprise an appropriate and common orthonormal set of basis vectors.
Eqs. (2.1.35) and (2.1.17) are both expressions for u. Hence
If we take the scalar products of the left-hand side and right-hand sides of eq. (2.1.40) with ![]() ,
, ![]() and
and ![]() , we obtain three linear equations, or a matrix equation of the form
, we obtain three linear equations, or a matrix equation of the form
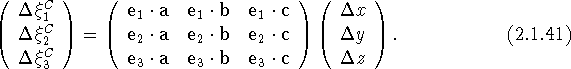
This is a transformation of the components of u, referred to the basis of the direct lattice, to its Cartesian components. The transformation matrix can be evaluated once the Cartesian basis vectors are defined (e.g., as above). If we adopt the index notation in the second line of eq. (2.1.17), eq. (2.1.41) can be written as
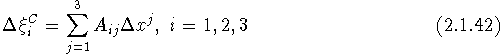
with
![]()
We can similarly transform a product of components of u between the lattice and Cartesian bases, and finally an average of such a product:
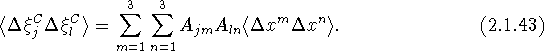
If we now make use of eqs. (2.1.22) and (2.1.37) we obtain
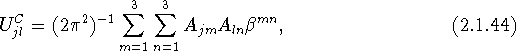
which is the expression for the transformation of ![]() to Cartesian coordinates.
to Cartesian coordinates.
The orthogonalization of U proceeds along similar lines. The required version of eq. (2.1.40) is now [cf. eq. (2.1.23)]
and following the same procedure by which eq. (2.1.41) is obtained, we arrive at the transformation
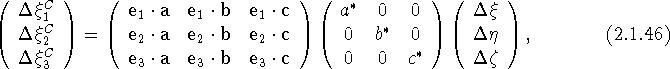
which relates the components of u, referred to the ![]() basis, to its Cartesian components. Equation (2.1.46) can be written concisely as
basis, to its Cartesian components. Equation (2.1.46) can be written concisely as
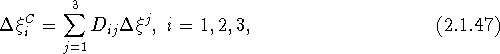
with
![]()
an element of the matrix product appearing in eq. (2.1.46). The desired transformation is obtained analogously to eq. (2.1.44) as
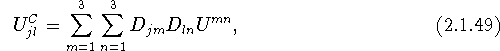
with ![]() and
and ![]() .
.
The explicit form of the transformation matrix appearing in eq. (2.1.41), for the specific Cartesian basis defined in eqs. (2.1.39), is:

Of course, a Cartesian basis associated with the direct and/or reciprocal bases can be chosen in an unlimited number of ways. A more general discussion of the construction of such Cartesian bases is given elsewhere (Shmueli, 1993).
2.2 Equivalent isotropic displacement parameters
It was pointed out by Hamilton (1959) and by Willis & Pryor (1975) that for minor departures from isotropic motion, or for anisotropic displacement parameters deemed to be physically insignificant, it may be worthwhile to replace the six-parameter description of anisotropic motion by a single quantity, which should describe an isotropic equivalent to the weakly or dubiously anisotropic case.
The IUCr Commission on Journals (1986) recommended that ``equivalent isotropic displacement parameters" be computed from the expressions proposed by Hamilton (1959) and by Willis & Pryor (1975). However, a number of different incorrect expressions have also been used (Fischer & Tillmanns, 1988), and this has led to considerable confusion. We first review the proper definitions and demonstrate their equivalence.
The first definition of the equivalent isotropic displacement parameter, as given by Hamilton (1959) and Willis & Pryor (1975), is
with ![]() an element of a mean-square displacement tensor, referred to a Cartesian basis [see eqs. (2.1.34) - (2.1.37)]. The trace of
an element of a mean-square displacement tensor, referred to a Cartesian basis [see eqs. (2.1.34) - (2.1.37)]. The trace of ![]() , as given on the right-hand side of eq. (2.2.51), is equivalent to the sum of the eigenvalues of this matrix. These eigenvalues are often computed, since an eigenvalue of the matrix
, as given on the right-hand side of eq. (2.2.51), is equivalent to the sum of the eigenvalues of this matrix. These eigenvalues are often computed, since an eigenvalue of the matrix ![]() represents the mean-square displacement along the corresponding eigenvector. The right-hand side of eq. (2.2.51) can then be interpreted as a mean-square displacement averaged over all directions.
represents the mean-square displacement along the corresponding eigenvector. The right-hand side of eq. (2.2.51) can then be interpreted as a mean-square displacement averaged over all directions.
Equation (2.2.51) can thus be applied to the computation of ![]() either by taking the trace of
either by taking the trace of ![]() , which was obtained from eq. (2.1.44) or eq. (2.1.49), or by using the sum of the eigenvalues of
, which was obtained from eq. (2.1.44) or eq. (2.1.49), or by using the sum of the eigenvalues of ![]() . However, it is essential to note that eq. (2.2.51) holds only for the Cartesian displacement tensor
. However, it is essential to note that eq. (2.2.51) holds only for the Cartesian displacement tensor ![]() . It will give incorrect values of
. It will give incorrect values of ![]() if U is referred to oblique basis vectors and its trace taken instead of that of
if U is referred to oblique basis vectors and its trace taken instead of that of ![]() .
.
Since the basis vectors of the Cartesian system have the property ![]() , a consideration of eqs. (2.1.35), (2.1.37), and (2.2.51) readily leads to
, a consideration of eqs. (2.1.35), (2.1.37), and (2.2.51) readily leads to
This equation is a convenient starting point for testing the equivalence of various definitions of ![]() . The second definition by Willis & Pryor (1975) is the first line of the next equation
. The second definition by Willis & Pryor (1975) is the first line of the next equation
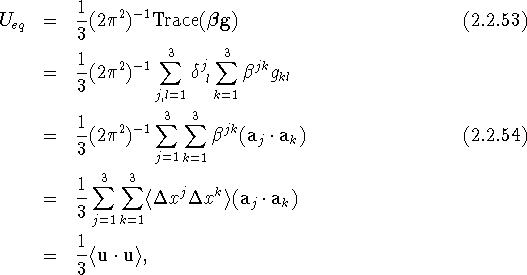
with g the real space metric tensor. This shows that eqs. (2.2.51) and (2.2.53), the two recommended definitions of ![]() , are equivalent.
, are equivalent.
If we make use of eqs. (2.2.54) and (2.1.38), two additional expressions for ![]() can be obtained:
can be obtained:
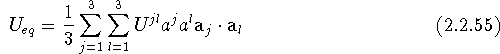
and
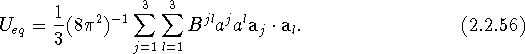
Thus, eqs. (2.2.51), (2.2.54), (2.2.55), and (2.2.56) are equivalent representations of the equivalent isotropic mean-square displacement parameter ![]() , obtainable from the commonly employed anisotropic displacement parameters.
, obtainable from the commonly employed anisotropic displacement parameters.
We can also arrive at eq. (2.2.54) by directly combining eqs. (2.1.44) and (2.2.51), and making use of a known property of the matrix A. We have
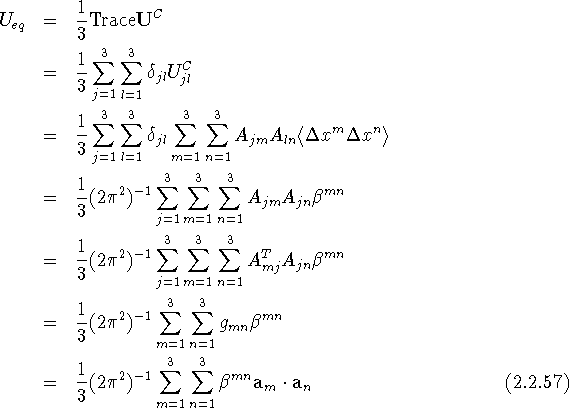
since
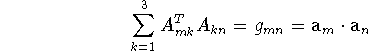
(e.g., Prince, 1982). This derivation shows that the value of ![]() does not depend on the particular form of the matrix A, which transforms the components of u from the lattice to the Cartesian basis.
does not depend on the particular form of the matrix A, which transforms the components of u from the lattice to the Cartesian basis.
Acta Crystallographica requires that published values of ![]() be accompanied by an evaluation of the standard deviation (now standard uncertainty) in these quantities. The calculation of this estimate is described in detail by Schomaker and Marsh (1983). A useful measure of the anisotropy of the mean-square displacement tensor is the ratio of its minimum and maximum eigenvalues. We recommend that published or deposited values of
be accompanied by an evaluation of the standard deviation (now standard uncertainty) in these quantities. The calculation of this estimate is described in detail by Schomaker and Marsh (1983). A useful measure of the anisotropy of the mean-square displacement tensor is the ratio of its minimum and maximum eigenvalues. We recommend that published or deposited values of ![]() be accompanied by both the standard uncertainties and the ratio of the minimum to the maximum eigenvalues of the corresponding anisotropic displacement tensors. Both the uncertainty of
be accompanied by both the standard uncertainties and the ratio of the minimum to the maximum eigenvalues of the corresponding anisotropic displacement tensors. Both the uncertainty of ![]() and the ratio may be helpful in judging the extent to which the use of
and the ratio may be helpful in judging the extent to which the use of ![]() is justified.
is justified.
2.3 Graphical representations of the Gaussian mean-square displacement matrix
Just as interatomic distances in crystals are most conveniently discussed in terms of atomic coordinates such as ![]() with dimension length rather than in terms of the dimensionless components
with dimension length rather than in terms of the dimensionless components ![]() , the physical interpretation of atomic displacement parameters is most convenient in terms of the mean-square displacement matrices U with elements having dimension (length)2 .
, the physical interpretation of atomic displacement parameters is most convenient in terms of the mean-square displacement matrices U with elements having dimension (length)2 .
2.3.1 Ellipsoids of constant probability
In the absence of anharmonicity, the anisotropic mean-square displacement matrix U can be regarded as the variance-covariance matrix of a trivariate Gaussian probability distribution with probability density function
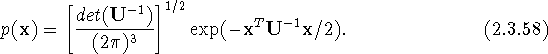
Here x is the vector of displacement of the atom from its mean position, and ![]() is the inverse of the quantity defined by eq. (2.1.25). If the eigenvalues of
is the inverse of the quantity defined by eq. (2.1.25). If the eigenvalues of ![]() are all positive, then the surfaces of constant probability defined by the quadratic forms
are all positive, then the surfaces of constant probability defined by the quadratic forms
are ellipsoids enclosing some definite probability for atomic displacement. This is the basis for the ORTEP ``vibration ellipsoids" (Johnson, 1965) that are used in so many illustrations of crystal structures. The lengths of the principal axes of the ellipsoids are proportional to the eigenvalues of the matrix ![]() expressed in the appropriate Cartesian system, and the directions of the principal axes correspond to the eigenvectors of this matrix. This representation cannot be used when
expressed in the appropriate Cartesian system, and the directions of the principal axes correspond to the eigenvectors of this matrix. This representation cannot be used when ![]() has one or more negative eigenvalues, because the resulting non-closed surfaces are no longer interpretable in terms of the underlying physical model.
has one or more negative eigenvalues, because the resulting non-closed surfaces are no longer interpretable in terms of the underlying physical model.
2.3.2 Mean-square displacement surface
The mean-square displacement amplitude (MSDA) in a direction defined by a unit vector n is
with n referred to the unit vectors ![]() ,
, ![]() , parallel to the reciprocal vectors
, parallel to the reciprocal vectors ![]() ,
, ![]() , respectively. The bases
, respectively. The bases ![]() and
and ![]() are mutually reciprocal (Hirshfeld & Rabinovich, 1966). Note that whereas
are mutually reciprocal (Hirshfeld & Rabinovich, 1966). Note that whereas ![]() in eq. (2.3.59) is dimensionless,
in eq. (2.3.59) is dimensionless, ![]() has dimension (length)2 . As n varies, the surface generated by
has dimension (length)2 . As n varies, the surface generated by ![]() is not an ellipsoid; it is usually peanut-shaped.
is not an ellipsoid; it is usually peanut-shaped.
Such surfaces can be constructed even for non-positive-definite tensors and they are therefore particularly useful for inspecting difference tensors ![]() between experimental
between experimental ![]() tensors and those obtained from kinematic or dynamic models of atomic and molecular motion (Hummel, Raselli & Bü, 1990).
tensors and those obtained from kinematic or dynamic models of atomic and molecular motion (Hummel, Raselli & Bü, 1990).
The distinction between the surfaces defined by eqs. (2.3.2) and (2.3.3) has often proved puzzling. Note that the right-hand side of (2.3.2) is a constant, the (arbitrarily chosen) equi-probability level for defining the ORTEP ellipsoids. When the matrix of the mean-square displacement tensor is non-positive definite, the quadratic surface defined by (2.3.2) is no longer closed, and no ellipsoid can be plotted. In contrast, the right-hand side of (2.3.3) is the mean-square displacement amplitude (MSDA) in a given direction, and varies as n varies. Only positive values of the quantity defined in (2.3.3) are meaningful for an individual atom, but negative values can be meaningful when differences in MSDA values are calculated. The MSDA surfaces can be plotted with the aid of the program PEANUT of Hummel et al. (1990); negative values are plotted as dashed contours.
3. Beyond the Gaussian Approximation
The situation is less straightforward if the distribution function is not Gaussian. A large variety of different approximation formalisms, as well as different nomenclature for similar formulations, is found in the literature. Summaries have been given by Johnson & Levy (1974), Zucker & Schulz (1982), Coppens (1993), and Kuhs (1992). By virtue of eq. (1.4.8), one may express either pdf(u) or ![]() as a series expansion and obtain the other quantity by Fourier transformation.
as a series expansion and obtain the other quantity by Fourier transformation.
The most widespread approaches are based on formalisms developed in statistics to describe non-Gaussian distributions (Johnson, 1969). They use a differential expansion of the Gausssian pdf. Two formulations are found in frequently used refinement programs, the cumulant or Edgeworth expansion ![]()
and the quasi-moment or Gram-Charlier expansion
with ![]() the Gaussian Debye-Waller factor (see sections 1.4 and 2.1) and
the Gaussian Debye-Waller factor (see sections 1.4 and 2.1) and ![]() ,
, ![]() , ... the third, fourth, ... order (anharmonic) tensorial coefficients. There are in general ten cubic, fifteen quartic, ... terms that enter into the treatment. In statistics they are called cumulants and quasi-moments, respectively. They constitute the parameters of the refinement. Various symbols for these coefficients are scattered through the literature. Greek letters are chosen here to comply with the
, ... the third, fourth, ... order (anharmonic) tensorial coefficients. There are in general ten cubic, fifteen quartic, ... terms that enter into the treatment. In statistics they are called cumulants and quasi-moments, respectively. They constitute the parameters of the refinement. Various symbols for these coefficients are scattered through the literature. Greek letters are chosen here to comply with the ![]() 's of the Gaussian case, which may thus be considered as second-order coefficients. For the same reason the factors
's of the Gaussian case, which may thus be considered as second-order coefficients. For the same reason the factors ![]() , with N the order of the tensor, are included, also to follow standard physical notation, which uses
, with N the order of the tensor, are included, also to follow standard physical notation, which uses ![]() as the scattering vector. The factors
as the scattering vector. The factors ![]() and/or the factors 1/N! (e.g., Kuhs, 1992) are sometimes omitted in the literature. For comparability of future results, it is therefore proposed that only coefficients defined as in (3.1.61) and (3.1.62) be published, and that subscripts be used to indicate the type of expansion employed.
and/or the factors 1/N! (e.g., Kuhs, 1992) are sometimes omitted in the literature. For comparability of future results, it is therefore proposed that only coefficients defined as in (3.1.61) and (3.1.62) be published, and that subscripts be used to indicate the type of expansion employed.
The ![]() ,
, ![]() , ... are dimensionless quantities. As proposed by Kuhs (1992), they may be transformed to quantities of dimension (length)N by
, ... are dimensionless quantities. As proposed by Kuhs (1992), they may be transformed to quantities of dimension (length)N by
with ![]() to be replaced by
to be replaced by ![]() ,
, ![]() ... Note that this is a generalization of eq. (2.1.38). It must be stressed, however, that the
... Note that this is a generalization of eq. (2.1.38). It must be stressed, however, that the ![]() ,
, ![]() , ... are simple expansion coefficients and (in general) have no direct physical meaning. The transformation (3.1.63) thus has no such merits as in the Gaussian case, and some real-space illustrations should always be given to permit the results to be appreciated. The best way is certainly to plot the corresponding pdf, obtained by inversion of (3.1.62) or (1.4.8). Only programs that produce sections of the pdf's seem to be currently available, although a three-dimensional visualisation similar to ORTEP would be highly desirable. Another way of presenting the results is by tensor contraction (Kuhs, 1992). For even-order terms, full contraction yields an invariant scalar,
, ... are simple expansion coefficients and (in general) have no direct physical meaning. The transformation (3.1.63) thus has no such merits as in the Gaussian case, and some real-space illustrations should always be given to permit the results to be appreciated. The best way is certainly to plot the corresponding pdf, obtained by inversion of (3.1.62) or (1.4.8). Only programs that produce sections of the pdf's seem to be currently available, although a three-dimensional visualisation similar to ORTEP would be highly desirable. Another way of presenting the results is by tensor contraction (Kuhs, 1992). For even-order terms, full contraction yields an invariant scalar,
For the Gram-Charlier series, this quantity indicates flatness (for negative values) or peakedness (positive values) of the pdf. The ![]() are the components of the real-space metric tensor. Note that
are the components of the real-space metric tensor. Note that ![]() , i.e., (3.1.64) is an extension of eq. (2.2.53). Similarly, vector invariants may be calculated for odd-order terms,
, i.e., (3.1.64) is an extension of eq. (2.2.53). Similarly, vector invariants may be calculated for odd-order terms,
giving the direction of maximal skewness. Partial contraction of even-order terms,
reveals the directions of flatness and peakedness.
Various discussions in the literature (see, e.g., Kuhs, 1992, and references therein) indicate that the Gram-Charlier formalism is the best choice in routine crystallographic work. In particular, it has the advantage that the reverse Fourier transformation (1.4.8) can be carried out analytically,
with ![]() Hermite polynomials, and
Hermite polynomials, and ![]() the harmonic part of the pdf. These polynomials are tabulated by Johnson & Levy (1974) up to the fourth order and by Zucker & Schulz (1982) up to the sixth order [see also Coppens (1993)]. The use of the Gram-Charlier expansion (3.1.62) is therefore recommended, although other formalisms may sometimes be advantageous for special problems. In any case, the results should always be carefully checked, especially if higher order terms are used merely to improve the agreement of the fit. Strong and extended negative regions in the pdf indicate inadequacy of the results. One also has to remember that, with anharmonic refinements, the positions and
the harmonic part of the pdf. These polynomials are tabulated by Johnson & Levy (1974) up to the fourth order and by Zucker & Schulz (1982) up to the sixth order [see also Coppens (1993)]. The use of the Gram-Charlier expansion (3.1.62) is therefore recommended, although other formalisms may sometimes be advantageous for special problems. In any case, the results should always be carefully checked, especially if higher order terms are used merely to improve the agreement of the fit. Strong and extended negative regions in the pdf indicate inadequacy of the results. One also has to remember that, with anharmonic refinements, the positions and ![]() obtained are not necessarily faithful representations of the mean and variance of the pdf, respectively. This must be borne in mind if bond distances and Gaussian displacement ellipsoids are to be derived from the refined parameters. In some situations, it may be better to use only the Gaussian approximation, even though the resulting R-factors may be higher.
obtained are not necessarily faithful representations of the mean and variance of the pdf, respectively. This must be borne in mind if bond distances and Gaussian displacement ellipsoids are to be derived from the refined parameters. In some situations, it may be better to use only the Gaussian approximation, even though the resulting R-factors may be higher.
![]()
In eqs. (3.1.61), (3.1.62), and many of the remaining equations in this section, the summation convention has been used. It is assumed that summation occurs over indices that are repeated, such as j, k, l and m in the terms on the right-hand side of ( 3.1.61 ) and ( 3.1.62 ).
Another possibility is the expansion of the so called one-particle-potential (OPP)
with Z the partition function. The second equality is obtained by setting ![]() .
.
The latter approach was formulated by Dawson & Willis (1967) and Willis (1969) for cubic point groups and later generalized for any symmetry by Tanaka & Marumo (1983). The OPP is written as
with ![]() the harmonic (quadratic) OPP and
the harmonic (quadratic) OPP and ![]() and
and ![]() the third and fourth order coefficients, respectively, which are defined in a Cartesian system. Since application of eqs. (3.1.68) and (1.4.8) does not lead to an analytical expression for
the third and fourth order coefficients, respectively, which are defined in a Cartesian system. Since application of eqs. (3.1.68) and (1.4.8) does not lead to an analytical expression for ![]() , the anharmonic part
, the anharmonic part ![]() is approximated in (3.1.68) by
is approximated in (3.1.68) by
The final expressions for ![]() are rather lengthy and may be found in Tanaka & Marumo (1983). Refinable parameters are the
are rather lengthy and may be found in Tanaka & Marumo (1983). Refinable parameters are the ![]() and
and ![]() . Other formulations with simpler expressions for
. Other formulations with simpler expressions for ![]() have been introduced by Coppens (1978), Kurki-Suonio, Merisalo & Peltonen (1979) and Scheringer (1985). None of these approaches seems to have been used much in crystallographic studies, and final recommendations must await further developments in this field. It should also be noted that the OPP approach treats each atom as an individual (Einstein-)oscillator, which is a poor approximation for tightly bound atoms in molecules.
have been introduced by Coppens (1978), Kurki-Suonio, Merisalo & Peltonen (1979) and Scheringer (1985). None of these approaches seems to have been used much in crystallographic studies, and final recommendations must await further developments in this field. It should also be noted that the OPP approach treats each atom as an individual (Einstein-)oscillator, which is a poor approximation for tightly bound atoms in molecules.
The OPP approach is physically meaningful only for purely dynamic displacive disorder (giving, e.g., the directions of weak and strong bonds), and is limited to rather small anharmonicities through the approximation (3.1.70). Occasionally special expansions (e.g., symmetry-adapted spherical harmonics) of pdf(u) or ![]() have been used for special problems (e.g., curvilinear motion, molecular disorder); see Johnson & Levy (1974), Press & Hüller (1973) and Prandl (1981). Again, these expansions do not seem yet to have entered routine crystallographic work. It should be remembered that the classical limit
have been used for special problems (e.g., curvilinear motion, molecular disorder); see Johnson & Levy (1974), Press & Hüller (1973) and Prandl (1981). Again, these expansions do not seem yet to have entered routine crystallographic work. It should be remembered that the classical limit ![]() , which is assumed in eq. (3.1.68), may be far from the actual situation even at room temperature.
, which is assumed in eq. (3.1.68), may be far from the actual situation even at room temperature.
4. Recommendations
- The term anisotropic displacement parameters (abbreviated ADPs) should be used in referring to the individual atomic coefficients in the exponent of the factor that describes the effects of atomic motion and static displacement.
- The elements of the tensors
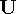 and
and 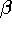 should always be superscripted when the refinement is referred to a crystal system rather than to a Cartesian system. This definition follows from the definition of the elements of and as contravariant tensor components (see Section 2.1, p. 11). The frequent use of subscripts for the ADPs, and specifically for those not referred to Cartesian systems, is inconsistent with their tensorial properties.
should always be superscripted when the refinement is referred to a crystal system rather than to a Cartesian system. This definition follows from the definition of the elements of and as contravariant tensor components (see Section 2.1, p. 11). The frequent use of subscripts for the ADPs, and specifically for those not referred to Cartesian systems, is inconsistent with their tensorial properties. - With the common Gaussian approximation, use either the quantities
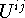 , which have dimension (length)2, defined in eq. (2.1.25), or the dimensionless
, which have dimension (length)2, defined in eq. (2.1.25), or the dimensionless 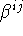 , defined in eq. (2.1.22).
, defined in eq. (2.1.22). - When the Gaussian approximation to the probability density function is not deemed valid, the use of the Gram-Charlier expansion of eq. (3.1.62) is recommended, although other formalisms may sometimes be advantageous for special problems.
- Standard uncertainties of ADPs obtained from a full-matrix refinement are valid within the system in which the refinement is made. If ADPs are transformed to any other axial system, Cartesian or not, then the uncertainties may also be calculated by transforming the original variance-covariance matrix to this new axial system and taking the square roots of its diagonal elements, i.e., the variances. The required variance-covariance matrix is usually not available for ADPs taken from the literature. Hence, although ADPs can still be transformed, their uncertainties cannot be. Calculations involving published ADPs and their (published) uncertainties should therefore be referred to the same system of coordinates as the original refinement in order to retain the significance of the published uncertainties.
- Avoid using the term ``temperature factor", both because the phenomenon represented may not be due entirely to thermal motion and because that phrase has in the past been used in several quite distinct senses (see Section 1.5).
- Avoid using the Gaussian anisotropic parameters that are now usually symbolized as
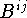 and are defined in eq. (2.1.26). These quantities are directly proportional to the recommended , the ratio being 8
and are defined in eq. (2.1.26). These quantities are directly proportional to the recommended , the ratio being 8 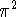 .
. - Avoid using ADPs that do not represent matrix elements. In some early references and computer programs, cross-terms were sometimes doubled in magnitude, being represented, for example, as
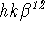 instead of
instead of 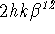 , for programming convenience. This was possible because the matrix representing the ADP is symmetric, with only six independent terms. This practice is not found in modern crystallographic software.
, for programming convenience. This was possible because the matrix representing the ADP is symmetric, with only six independent terms. This practice is not found in modern crystallographic software. - Published values of
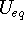 should always be accompanied by their standard uncertainties. The ratio of the minimum to the maximum eigenvalues of the corresponding anisotropic displacement tensors should also be published, either in the primary publication itself or in the secondary (deposition) publication.
should always be accompanied by their standard uncertainties. The ratio of the minimum to the maximum eigenvalues of the corresponding anisotropic displacement tensors should also be published, either in the primary publication itself or in the secondary (deposition) publication.
Authors of crystallographic software and crystallographers who maintain their own software are encouraged to introduce the minor modifications that are required for the implementation of these Recommendations.
References
- 1
- COPPENS, P. (1978). In Electron and Magnetization Densities in Molecules and Crystals, P. Becker, ed., pp. 521-543. New York: Plenum Press.
- 2
- COPPENS, P. (1993). In International Tables for Crystallography, Vol.B: Reciprocal Space, U. Shmueli, ed., Section 1.2.11. Dordrecht: Kluwer Academic Publishers.
- 3
- COPPENS, P. & BECKER, P.J. (1992). In International Tables for Crystallography, Vol.C: Mathematical, Physical and Chemical Tables, A.J.C. Wilson ed., Chapter 8.7. Dordrecht: Kluwer Academic Publishers.
- 4
- COWLEY, J.,M. (1992). In International Tables for Crystallography, Volume C: Mathematical, Physical and Chemical Tables, ed. A.J.C. Wilson, pp. 223-245. Dordrecht: Kluwer Academic Publishers.
- 5
- CRUICKSHANK, D. W. J. (1956). Acta Cryst. 9, (a) 747-754; (b) 754-757; (c) 757-758; (d) 915-923; (e) 1005-1009; (f) 1010-1011.
- 6
- DAWSON, B. & WILLIS, B.T.M. (1967). Proc.R.Soc.London Ser.A 298, 307-315.
- 7
- DEBYE, P. (1913). Verh. Dtsch. Phys. Ges. 15, 738-752.
- 8
- DUNITZ, J.D., SCHOMAKER, V. & TRUEBLOOD, K.N. (1988). Journal of Physical Chemistry 92, 856-867.
- 9
- FISCHER, R.X. & TILLMANNS, E. (1988). Acta Cryst. C44, 775-776.
- 10
- HAMILTON, W.C. (1959). Acta Cryst. 12, 609-610.
- 11
- HIRSHFELD, F.L. & RABINOVICH, D. (1966). Acta Cryst. 20, 146-147.
- 12
- HUMMEL, W., RASELLI, A. & BURGI, H.-B. (1990). Acta Cryst. B46, 683-692.
- 13
- IUCR COMMISSION ON JOURNALS (1986). Acta Cryst. C42, 1100.
- 14
- JAGODZINSKI, H. & FREY, F. (1993). In International Tables for Crystallography, Vol.B: Reciprocal Space, U. Shmueli, ed., Chapter 4.2. Dordrecht: Kluwer Academic Publishers.
- 15
- JOHNSON, C.K. (1965). A Fortran Thermal Ellipsoid Plot Program for Crystal Structure Illustrations. ORNL report no. 3794. Oak Ridge National Laboratory, Oak Ridge, Tennessee.
- 16
- JOHNSON, C.K. (1969). Acta Cryst. A25, 187-194.
- 17
- JOHNSON, C.K. & LEVY, H.A. (1974). In International Tables for X-ray Crystallography, Vol. IV, pp.311-336. Birmingham: Kynoch Press.
- 18
- KENDALL, M. & STUART, A. (1977). The Advanced Theory of Statistics, Vol 1, 4th ed. London: Griffin.
- 19
- KUHS, W.F. (1992). Acta Cryst. A48, 80-98.
- 20
- KURKI-SUONIO, K. MERISALO, M. & PELTONEN, H. (1979). Phys. Scr. 19, 57-63.
- 21
- MASLEN, E.N., FOX, A.G. & O'KEEFE, M.A. (1992). In International Tables for Crystallography, Vol.C: Mathematical, Physical and Chemical Tables, A.J.C. Wilson ed., Section 6.1.1. Dordrecht: Kluwer Academic Publishers.
- 22
- PRANDL, W. (1981). Acta Cryst. A37, 811-818.
- 23
- PRESS, W. & HÜLLER, A. (1973). Acta Cryst. A29, 252-256.
- 24
- PRINCE, E. (1982). Mathematical Techniques in Crystallography and Materials Science. New York: Springer-Verlag.
- 25
- SCHERINGER, C. (1985). Acta Cryst. A41, 73-79.
- 26
- SCHOMAKER, V. & MARSH, R. E. (1983). Acta Cryst. A39, 819-820.
- 27
- SCHWARZENBACH, D., ABRAHAMS, S.C., FLACK, H.D., PRINCE, E. & WILSON, A.J.C. (1995). Acta Cryst. A51. 565-569.
- 28
- SHMUELI, U. (1993). In International Tables for Crystallography, Volume B: Reciprocal Space, ed. U. Shmueli, pp. 2-9. Dordrecht: Kluyver Scientific Publications.
- 29
- SPAIN, B. (1956). Tensor Calculus. Second edition, Chap. I. London: Oliver and Boyd.
- 30
- TANAKA, K. & MARUMO, F. (1983). Acta Cryst. A39, 631-641.
- 31
- VAINSHTEIN, B.,K. (1964). Structure Analysis by Electron Diffraction. Oxford: Pergamon Press.
- 32
- VAINSHTEIN, B.K. & ZVYAGIN, B.B. (1993). In International Tables for Crystallography, Volume B: Reciprocal Space, ed. U. Shmueli, pp. 310-314. Dordrecht: Kluwer Academic Publishers.
- 33
- WALLER, I. (1923). Z. Phys. 17, 398-408.
- 34
- WILLIS, B.T.M. (1969). Acta Cryst. A25, 277-300.
- 35
- WILLIS, B.T.M. & PRYOR, A.W. (1975). Thermal Vibrations in Crystallography. Cambridge: Cambridge University Press.
- 36
- WILLIS, B.T.M. (1993). In International Tables for Crystallography, Vol.B: Reciprocal Space, U. Shmueli, ed., Chapter 4.1. Dordrecht: Kluwer Academic Publishers.
- 37
- WILSON, E.B., DECIUS, J.C. & CROSS, P.C. (1954). Molecular Vibrations. New York: McGraw-Hill
- 38
- ZUCKER, U.H. & SCHULZ, H.H. (1982). Acta Cryst. A38, 568-576.
These pages are maintained by the Commission Last updated: 15 Oct 2021