
Meeting report
Protein Society Symposium
The Eighth Symposium of the Protein Society was held July 9-13, 1994 in San Diego, CA, USA. The annual Symposium brings together all aspects of protein structure determination and functional analysis and, of necessity, includes a major emphasis on data from X-ray crystal structure analysis. Nearly one third of the oral presentations were from leaders in the crystallographic community and a similar proportion of the 550 posters described macromolecular crystal structure determinations.
A very prominent theme in these crystallographic reports concerned the relevance of molecular flexibility to biological activity and the ability to gain reliable information about that flexibility from carefully designed studies of macromolecular structure in different crystals and conformational states.
P. Sigler illustrated a remarkable three switch sequence of structural changes in Guanadine triphosphatase (GTPase) that accompanies the transition from GDP to GTP binding. The dynamic changes that he postulates on the basis of structure studies of the enzyme containing GDP and GTP are analogous to the meshing of gears in an elaborate clock and have direct implications concerning enzyme action. In her elaborate description of the highly complex, but increasingly well understood, glycogen phosphorylase system, L. Johnson drew attention to a dramatic shift in a peptide chain accompanying an active to inactive transition in a phosphorylase enzyme. She showed that the reorganization of the chain that accompanied serine phosphate complexation produced a 60 Å shift in the position of the chain terminal. This major shift in the conformation of the long chain is related to transformation of the electronic nature of the surface of the enzyme as it goes from an active to an inactive (R and T) state.
P. Kim, recipient of the Young Investigator Award, sponsored by DuPont Merck Pharmaceutical Co., presented a brilliant lecture on 2, 3, and 4 strand coiled coils that drew upon the X-ray studies by T. Albers and others. The highlight of his talk also concerned a major rearrangement in protein conformation.
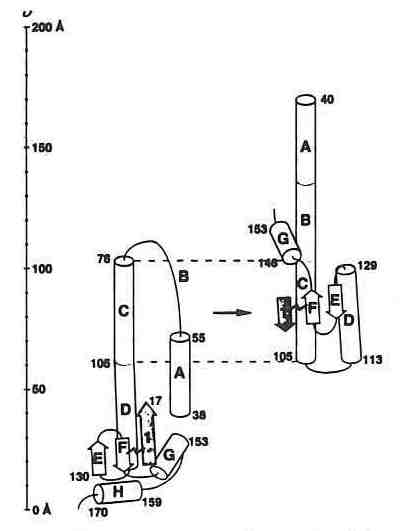 Schematic illustration of a conformational change in the influenza virus hemagglutinin which mediates fusion of the viral and host cell membrane. Taken from a paper by Wiley and co-workers on the structure of a fragment of hemagglutinin that revealed a major refolding of the structure (Nature 371, 37-43, 1994).
Schematic illustration of a conformational change in the influenza virus hemagglutinin which mediates fusion of the viral and host cell membrane. Taken from a paper by Wiley and co-workers on the structure of a fragment of hemagglutinin that revealed a major refolding of the structure (Nature 371, 37-43, 1994).
In a virus of known structure, a fusion peptide that must interact with a membrane receptor is buried 100 Å beneath the viral surface where critical membrane interaction is presumed to take place. Kim noted that this fusion peptide is at the end of a protein strand that has a sequence suited to helical formation and to coiled coil formation. He predicted conditions under which the formation of the coiled coil would be triggered causing the fusion protein to undergo a 100 Å position shift placing it in position to bind to the membrane receptor. Kim supported his theoretical model with an illustration from a recent X-ray structure determination of the relevant coiled coil fragment in hemagglutinin from Don Wiley's lab (see figure) that postulates a spring loaded mechanism for flu virus infection. In a chilling footnote, Kim cautioned that if this spring loaded element is a characteristic of virus structure, the design of vaccines based upon virus fragments or peptide mimics could trigger the activation of the virus rather than its suppression. In the case of the hemagglutinin, the firing of the spring and repositioning of the fusion peptide occurs after the shift or removal of protein domains at the end of probes that extend from the virus surface.
M. Rossmann received the Stein and Moore Award sponsored by the Merck Foundation. A session in his honor featured talks on virus structure including presentations by S. Harrison and Rossmann. Another question concerning the critical translocation of a protein in a macromolecular structure concerned the mechanism by which VP4, a protein buried deep within a virus, gets out of the dodecahedron without disrupting its integrity. Rossmann postulates that VP4 might escape through the channel surrounding the fivefold axis once an ion plug in the channel is removed. Rossmann has previously identified the canyons on viral surfaces where antiviral agents have been shown to bind. Recently he has used electron microscopy to show that cell surface adhesion molecules also bind in these canyons as a probable means of cell attachment by viruses.
C. Pabo described the rapidly expanding number of structure determinations of various DNA binding proteins. In certain types, general conformation and recognition seem to accommodate extensive sequence variation. These observations are consistent with other studies in which an overall similarity in protein fold exists with little or no relation to conservation of amino acid sequence.
 N. Pavletich determined the crystal structure of p53, the protein Science labeled the molecule of the year 1993. (Photo WLD)
N. Pavletich determined the crystal structure of p53, the protein Science labeled the molecule of the year 1993. (Photo WLD)
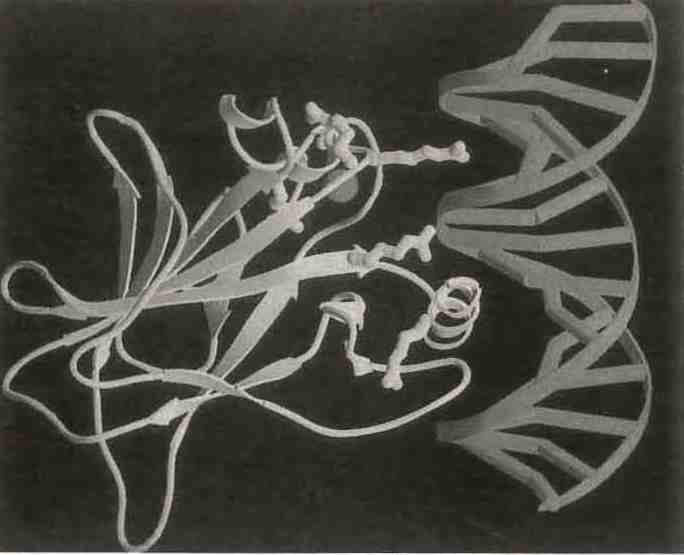 The 3D structure of p53, an enzyme implicated in a wide range of cancers, only deepens the mystery surrounding its behavior (Y. Cho et al., Science 265, 346-355, 1994).
The 3D structure of p53, an enzyme implicated in a wide range of cancers, only deepens the mystery surrounding its behavior (Y. Cho et al., Science 265, 346-355, 1994).
The structure of p53, named Molecule of the Year 1993 by Science, presented a dramatic and puzzling counterexample to that general rule. It has been discovered that 80% of all tumors have mutationsin p53, a protein that is intimately involved in apoptosis (cell death). T. Jacks described studies of mutant forms of p53 that indicate that almost any changes made to the 200 amino acids in the center of the 339 amino acid protein will inactivate it, presumably by altering overall conformation and impairing DNA interactions. N. Pavletich presented the details of his crystal structure determination of p53 with a fragment of DNA. The determination allows unequivocal identification of the location of mutational changes that alter the activity of p53. It is not immediately apparent why this protein should be so intolerant to any sequence variation when so many other protein conformations are retained with little or no sequence homology at all.
Among the posters there were other interesting examples of significant conformational change observed in different crystal forms, including the well established case in the serpin family where an inhibitory loop that extends away from the bulk of the protein in one form threads neatly into a β sheet in the interior of the protein in the other (M. R. Wardell, Cambridge, UK). In another interesting example, a single mutation buried in the active cleft of glutamate dehydrogenase causes an opening of the jaws around the cleft that is much greater than the change observed upon addition of NAD cofactor and/or inhibitors.
In the structure of a tyrosine phosphatase (H. I. Schubert, U. Michigan, USA), a residue that is shown by mutation studies to be critically important to activity, even though it is not in the active site, moves by 9 Å in the crystal structure of the ligated protein in comparison to the unligated.
One of the most active areas of protein research is the investigation of folding mechanisms. A remarkable determination that circularly permuted aspartate transcarbamolase (ATCase, L. Gonzalez, U. California, Berkeley, USA) will fold the same way that native ATCase does may help answer some questions and raise new ones. When the crystal structure determination revealed that the carbonyl and amino of ATCase are in close proximity (a not uncommon phenomenon) a mutant form was engineered by joining the wild type N- and C- termini and splicing it in the middle creating new ends.
Other signs of the impact of X-ray crystallography upon the Protein Society include the election of B. Mathews as president for 1995-96 and the presence of Enraf Nonius and Hampton Research in the Exhibition Hall. The 1995 meeting will be in Boston, July 8-12 and the 1996 meeting will again be in San Diego, August 3-7, the week before the IUCr Congress in Seattle. Protein crystallographers from abroad may want to consider extending their stay in the US to include attendance at the San Diego meeting 1996.
There seems to be no end in sight for the surprises that nature has in store for us if we keep examining the structural variations possible by modulating sequence, cofactors, solvents, substituents, and crystal growth conditions in our exploration of molecular structures via crystallography, the most reliable source of data on stable molecular conformation.
W. L. Duax