
Feature article
Structure-Based Drug Design
 Michael Rossmann and Louise Johnson, Plenary lecturers at the IUCr Congress in Beijing, discuss structure-based drug design after Louise's talk. (Photo WLD)
Michael Rossmann and Louise Johnson, Plenary lecturers at the IUCr Congress in Beijing, discuss structure-based drug design after Louise's talk. (Photo WLD)
The earliest known drugs were those derived from natural products whose beneficial effects were detected from observation. Such compounds include aspirin, acetylsalicylate. The salicylates were originally detected in the bark of willows (Salix) and in the plant meadow sweet (Spiraea ulmaria), from which the name comes. The organic synthesis was achieved 100 years ago. Today nearly 40 million tons of aspirin are sold per year and it is widely used from the relief of headaches to the prevention of heart attacks. In 1971 John Vane proposed that the action of aspirin derived from its ability to inhibit prostaglandin biosynthesis. The three-dimensional structure of the target enzyme, prostaglandin H2 synthase-1, has now been solved by Michael Garavito and his colleagues (Nature, 1994, 367, 243-249). The structural work has shown how acetylation of a particular serine by aspirin could block access to the cycloxygenase catalytic site and inhibit the enzyme. While a complete understanding of aspirin's anti-inflammatory activity requires the structure of the related isoform prostaglandin synthase-2, we now have a beautiful example of the working of one of the most successful and yet the most simple pharmaceutical agents of all time. Is it possible to go the other way - from protein structure to drug?
The conceptual breakthrough, developed in the nineteenth century, that a drug has a highly specific target at the cellular level led to the notion of receptor-based drug design that has profoundly benefited the drug discovery process. The strategy in the design of new agents has been to modify the structure of a lead compound by systematic modification of its chemical structure in conjunction with biological and physiological evaluation. The discovery of the drug cimetidine for the treatment of duodenal ulcers by James Black and colleagues provides an example. The coordinated program encompassed clinical experience, pharmacological modeling, and chemical synthesis. It was known that histamine promoted the secretion of acid and that histamine also had a role in the allergic response. It was deduced that there should be two receptors H1 and H2 that were stimulated by the same agent, histamine. To block acid secretion only one receptor, the H2 must be hit. Starting with histamine, the molecule was modified first on the ring which proved nonproductive and then on the side chain that led to cimetidine. Although much was known of the active agent, histamine, and its physiological effects, nothing was known of the structure of the target molecule, the histamine H2 receptor.
The most recent phase in drug design has utilized knowledge of the structure of target molecules or of related compounds. Knowledge of the three-dimensional structures of pharmacologically important receptor-ligand complexes has the potential for profoundly influencing and accelerating the discovery and development of lead compounds for drug design. From experimental observations at the atomic level of how inhibitors bind to their macromolecules, specific interactions that are important in molecular recognition can be inferred. This knowledge can be applied to lead compounds in de novo design as well as to improvement in existing leads and can provide insights into mechanisms of existing drugs. This methodology is now being applied in a systematic fashion to define new products.
One of the earliest and best known examples of this approach is the use of knowledge of the carboxyopeptidase structure to design Captopril, a potent inhibitor of the angiotensin converting enzyme. Later knowledge of thermolysin was also used to design and test further potent compounds. These compounds have become some of the most valuable drugs for treating hypertension and heart failure and their annual sales total more than $6B. However, although much attention has been focused on the potential of structure-based drug design the number of definitive compounds that have neared the market place is limited. Here I select four examples where there is a reasonable expectation of success.
Influenza virus neuraminidase inhibitors
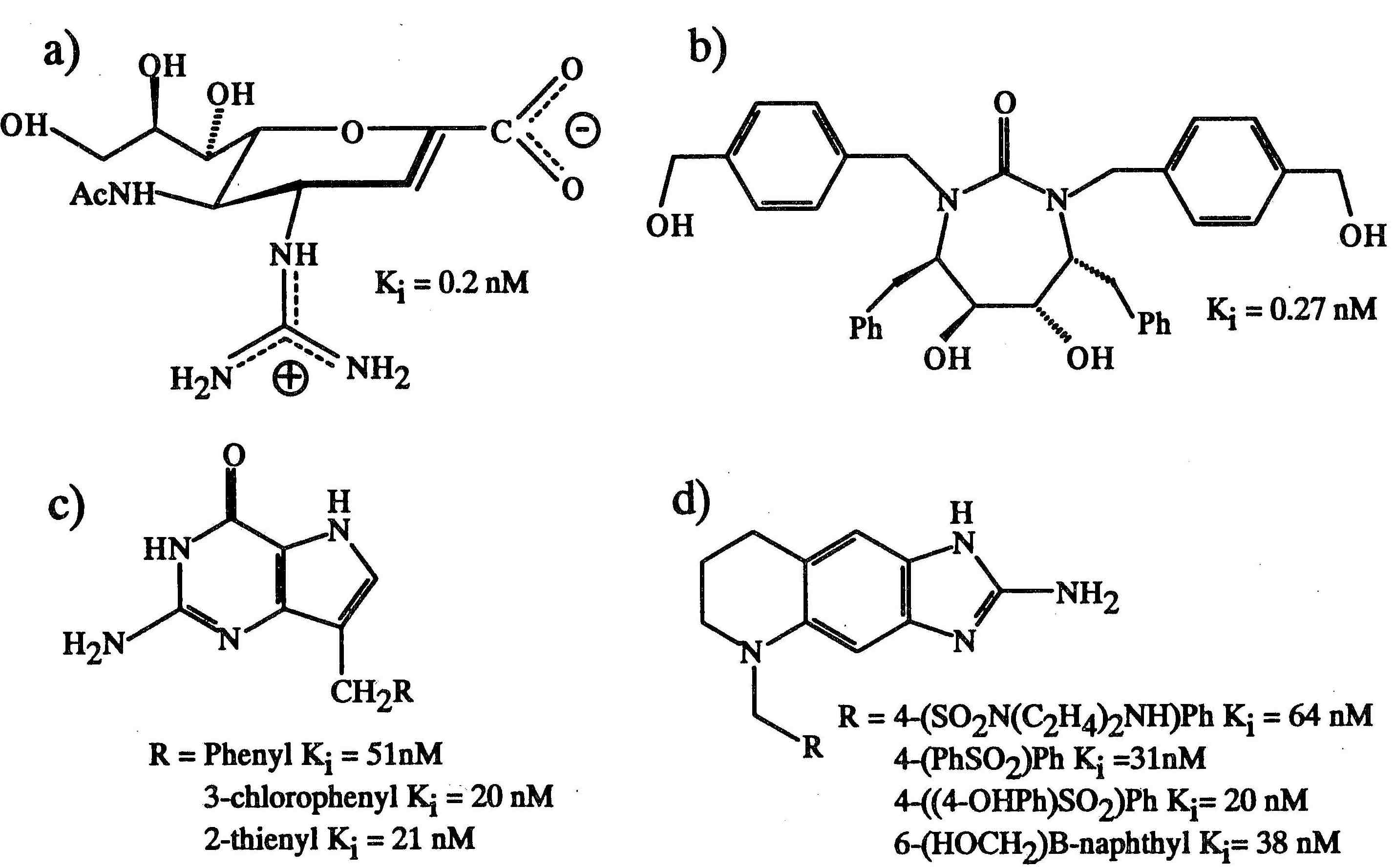 Inhibitors of (a) influenza virus neuraminidase, (b) HIV protease, (c) purine nucleoside phosphorylase, and (d) thymidylate synthase designed using X-ray crystallographic information. (Photo courtesy of MFB Graphics)
Inhibitors of (a) influenza virus neuraminidase, (b) HIV protease, (c) purine nucleoside phosphorylase, and (d) thymidylate synthase designed using X-ray crystallographic information. (Photo courtesy of MFB Graphics)
Neuraminidase, the surface bound enzyme of the influenza virus, cleaves sialic acid from glycoproteins. The enzymatic action probably assists in the elution of newly synthesized virions from infected cells and in the passage of the virus through the respiratory tract mucus. Researchers in Australia (Von Itzstein et al., Nature, 1993, 363, 418-423) have used knowledge of the structure of the enzyme complexed with a transition state compound and the computer program GRID (Wade et al., J. Med. Chem., 1993, 36, 140-156) to design new compounds. The addition of a guanidino group to the 4 position of the unsaturated neuraminic acid, 2-deoxy-2,3-didehydro-D-N-acetylneuraminic acid, allowed interactions with two carboxylates. The new compound [Figure 1 (a)] exhibited a Ki=0.2 nM, four orders of magnitude better than the starting compound, and scientists at Glaxo, UK have shown that it has significant in vivo activity against influenza virus replication. The agent is in clinical trials and can be administered as a mouth spray.
HIV protease
In the search for new therapeutic compounds to combat AIDS, the HIV protease is probably the protein most widely studied by X-ray methods. It has been estimated that there are at least 140 structure determinations of inhibited complexes of HIV protease and yet impressive results continue to appear. The latest of these is reported in a recent paper in Science by Lam, Erickson-Vitanen and colleagues at the DuPont Merck Pharmaceutical Company (Science, 1994, 263, 380-384). Mechanistic information was used in the design of inhibitors that avoided the use of peptide-based molecules that have poor oral bioavailability, exploited the two-fold symmetry of the dimeric enzyme with a C2-symmetric diol compound to target the two aspartates, included the use of a cyclic urea carbonyl oxygen to mimic the hydrogen bond features of a key structural water molecule, and engineered selectivity restrained by a preorganized scaffold from knowledge of the binding site geometry. The result was a series of relatively low molecular weight compounds with high oral bioavailability and good potency against the virus. Compound DMP 323 [Figure 1(b)] has a Ki=0.27 nM and is currently undergoing phase I clinical investigation.
Purine nucleoside phosphorylase
Purine nucleoside phosphorylase is a salvage enzyme important to the T-cell mediated part of the immune system. Its interest as a drug target arises from two rather different properties. The enzyme metabolizes purine nucleosides, including anticancer agents such as 2'-deoxy-6-thioguanosine and the anti-AIDS drug 2',3'-dideoxyinosine and hence its inhibition might allow these drugs longer action. Second, inhibitors may have application in the treatment of T-cell proliferative diseases, such as T-cell lymphoma, or in the suppression of host vs graft response in transplant operations. However, the design of inhibitors is made difficult by the need to ensure high membrane permeability of the compounds in addition to specific and potent inhibition in competition with substrate phosphate. Starting with the observation that 5'-deoxy-5'-iodo-9-deazainosine was the most potent available inhibitor and that the N-9 position allowed substitution of groups, Montgomery et al. (J. Med. Chem. 1993, 36, 55-69) report the design and synthesis of a number of 9-(arylmethyl) derivatives of 9-deazaguanine [Figure l(c)] using the three-dimensional structure of the human enzyme, X-ray analysis of inhibitor complexes, and Monte Carlo methods to predict likely compounds. The compounds have IC50 values of the order of 20 nM at low phosphate and 200 nM at high phosphate (50 mM). The improved potency of the 9-substituted compounds has been rationalized from X-ray analysis where subtle changes in hydrogen bonding patterns and location of the phenyl group between two phenylalanine side chains were observed. It has been reported that phase I clinical trials for some of these compounds have been completed.
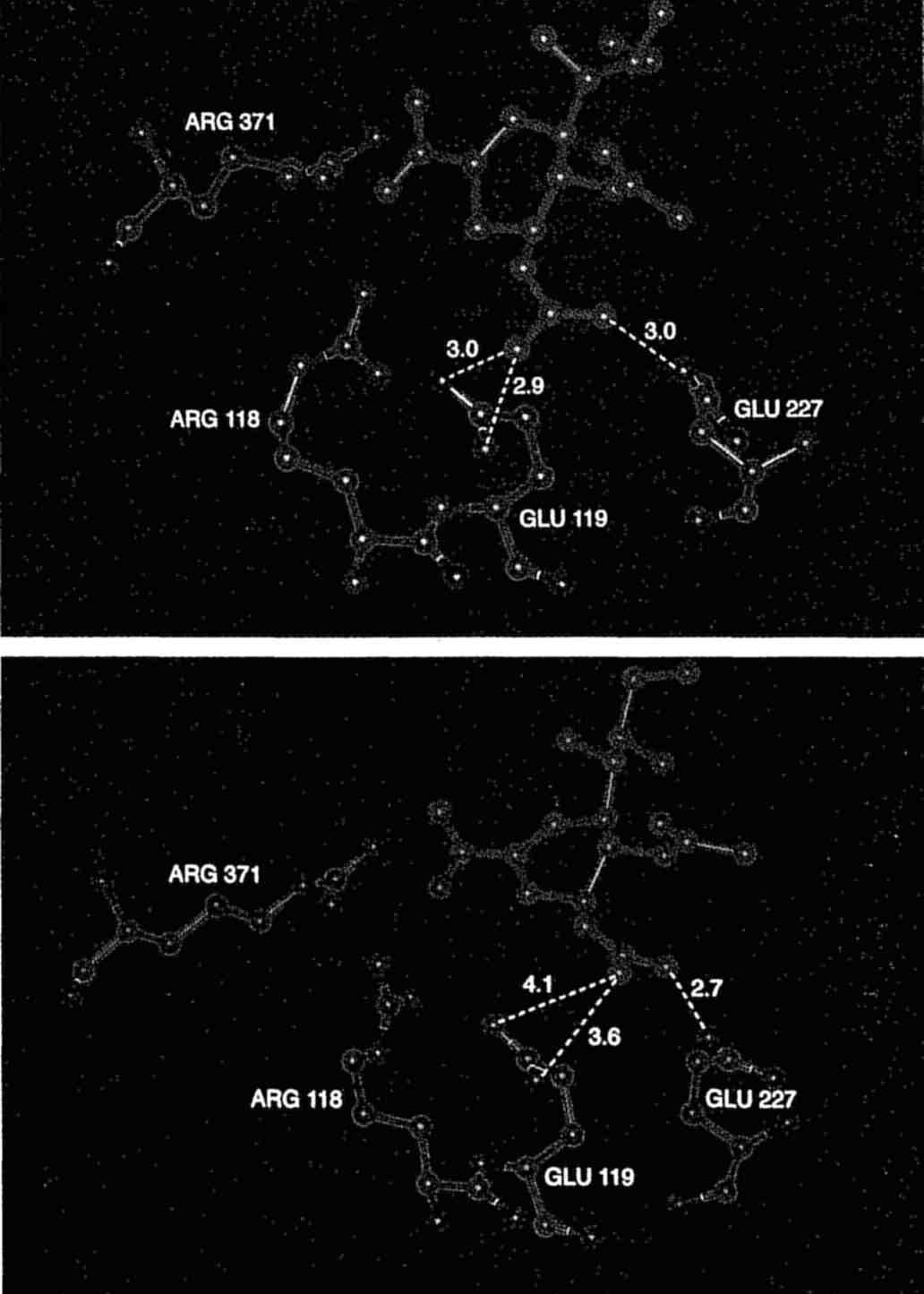 A comparison of the predicted (top) and observed interactions of two inhibitors of influenza virus sialdose. (Photo courtesy of M. Von Itzstein)
A comparison of the predicted (top) and observed interactions of two inhibitors of influenza virus sialdose. (Photo courtesy of M. Von Itzstein)
Thymidylate synthase
Thymidylate synthase is the limiting enzyme in the metabolic pathway for the de novo synthesis of thymidylate and as such is a target for drugs against cancer and other proliferative cell diseases. Inhibitors that compete with the substrate deoxyuridylate monophosphate, such as 5-flourouracil, already exist and are used as clinical agents. There is an interest in developing inhibitors against the folate cofactor to provide compounds that do not require further metabolism into the active agent and avoid toxicity problems sometimes associated with nucleotide-based inhibitors. Using the E. coli structure of thymidylate synthase as a model for the human enzyme (70% identity in sequence), Reich et al. (J. Med. Chem., 1992, 35, 847-858) have developed a series of 6,7-imidazotetrahydroquinoline inhibitors that have improved inhibition constants from micromolar range to 15 nM [Figure l(d)]. The program GRID was used to map the space available for ligands. A crystal structure of the first test compound (Ki=7μM) used to engineer new hydrogen bonds revealed hydrophobic interactions and the displacement of a water molecule. Some of the resulting compounds were shown to effectively inhibit the growth of three tumor cell lines in vitro. Curiously, it appeared that cell growth inhibition may not have been the primary consequence of thymidylate synthase inhibition. As the authors discuss, the information from a protein crystal structure has the potential of increasing selectivity for a given target, but having a tight inhibitor does not necessarily result in the specific targeting of that enzyme in cells. Factors which are difficult to predict, such as binding to other cellular proteins, metabolism, or transport properties can render a potent enzyme inhibitor either ineffective or possibly more effective in cell culture and in vivo.
A selection of proteins where structural knowledge has contributed to the design of new therapeutic agents and/or an understanding of the action of existing agents.
| PROTEIN | DISEASE | INHIBITOR/DRUG |
|---|---|---|
| Haemoglobin | Sickle cell anaemia | Salicylaldehyde compounds, clofibric acid, bezafibrate |
| Dihydrofolate Reductase | Cancer | Methotrexate |
| Thymidylate Synthase | Cancer | Flourouracil and 6,7-imidazotetrahydroquinoline compounds |
| Nucleoside Phosphorylase | Cancer | 9-(arylmethyl) derivatives of 9-deazaguanine |
| Angiotensin Converting Enzyme | Hypertension | Captopril and enalopril |
| Renin | Hypertension | Peptide-based transition state analogs |
| Human Leucocyte Elastase | Degenerative Lung Disease | α1-antitrypsin, flouromethyl ketones and cephalosporin analogs |
| Thrombin | Anticoagulant therapy | Hirudin and noncleavable hirudin analogs; argatroban |
| Tissue Plasminogen Activator Inhibitor | Anticoagulant therapy | Engineered recombinant protein to prevent inhibition |
| Carbonic anhydrase | Ocular hypertension, glaucoma | Thienothiopyran-2-sulfonamides (MK507) |
| DNA Gyrase | Bacterial infection | Quinolones (Nalidixic acid), Coumarins (Novobiocin) |
| Adenosine Deaminase | Severe Combined Immune Deficiency disease | Gene therapy |
| HIV Protease | AIDS | C2 symmetric diols based on seven membered cyclic urea scaffold; peptide-based transition state analogs |
| HIV Reverse Transcriptase | AIDS | AZT, nevirapine |
| Human Rhino Virus | Common Cold | Sterling-Winthrop Pharmaceutical Research Division and Janssen Research Foundation compounds |
| Influenza Virus Neuraminadase | Influenza | Modified neuraminic acid analog |
In addition to the above examples, there has been a host of structural results on proteins of medical importance. Some of these are summarized in the Table. They include examples that contribute to knowledge of how existing agents work and where knowledge of the structure can lead to new compounds with improved properties. Examples include work with acetylcholinesterase for agents that block neurotransmission, with bacterial DNA gyrase for understanding the action of some antibiotics, or with HIV reverse transcriptase for understanding the mechanism of AZT. Structural data have led to clues for improvement in properties of the antiviral compounds against human rhino virus, the causative agent of the common cold. The structure of the cyclophilin A-cyclosporin complex is of key importance for understanding the immunosupressant activity of the drug cyclosporin. Work in my own laboratory with glycogen phosphorylase has exploited knowledge of the structure and regulatory properties of this large enzyme to design glucose-analog inhibitors that may be of importance for the regulation of hepatic glycogen metabolism and blood glucose levels in diabetic patients. Understanding protein structure has led to new proteins produced by recombinant DNA technology such as a fast acting insulin for treatment of diabetes or "humanized" antibodies for the treatment of leukemia and arthritis. Finally, new results on the human histocompatibility complex and associated bound peptides, of cytokines and cytokine receptor complex, superantigens, the human growth hormone, and its complex with its receptor have provided new insights into basic recognition events in the immune response, the inflammatory response, and hormone response.
 Chong-Hwan Chang, seen here organizing his transparencies for a presentation at the Pittsburgh Diffraction Conference in Philadelphia, November 3-5, 1993, prepared the cover illustration of the cyclic urea in the HIV protease binding site. (Photo WLD)
Chong-Hwan Chang, seen here organizing his transparencies for a presentation at the Pittsburgh Diffraction Conference in Philadelphia, November 3-5, 1993, prepared the cover illustration of the cyclic urea in the HIV protease binding site. (Photo WLD)
Despite the immense wealth of information and the recent positive results that have come from the structural knowledge, structure-based drug design is still in its infancy. The design of tight inhibitors has been impressively accomplished but the more stringent requirements that a drug must fulfill in order to treat patients such as bioavailability, toxicity, metabolism, and half-life are more difficult to address. Structure-based drug design has a long way to go before it can rival the products such as cortisones, antibiotics, antidepressants, hypoglycaemic agents that were produced by more conventional methods but it provides an alternative approach. It has been estimated (Vagelos, Science, 1991, 252, 1080-1084) that on average in a trial and error procedure some 10,000 compounds will be screened, 10 will go forward to trials and 1 may become a prescription medicine. Seven out of ten medicines do not recoup their cost and fewer than five earn more than $1B per year. There are diseases of the rich and diseases of the poor and the majority of life-threatening diseases occur in poor countries where the likelihood of regaining costs is small. Yet there are encouraging signs such as the donation by the Merck company of ivermectin, a potent anti parasite agent, for the treatment of river blindness. Many diseases are a product of poor social and health conditions. The development of cures lies also in improving these conditions. It is to be hoped that the new knowledge created by crystallography will also contribute.
I am grateful to P. J. Goodford for comments on this manuscript.
Louise Johnson