
Feature article
Synchrotron radiation: new opportunities for chemical crystallography
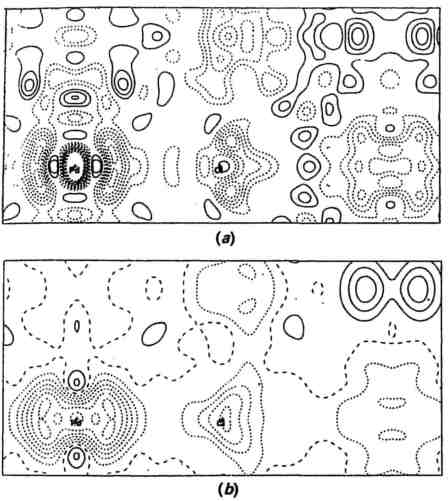 Fig. 1 Difference electron density maps for the (110) plane of K2PdCl4. (a) Is calculated from MoKα data and (b) is calculated from SR data recorded at Photon Factory, Japan, using a wavelength of 0.7 Å. [From Hester, Maslen, Spadaccini, Khiziwa and Satow (1993)]
Fig. 1 Difference electron density maps for the (110) plane of K2PdCl4. (a) Is calculated from MoKα data and (b) is calculated from SR data recorded at Photon Factory, Japan, using a wavelength of 0.7 Å. [From Hester, Maslen, Spadaccini, Khiziwa and Satow (1993)]The first dedicated sources of synchrotron radiation (SR) came into operation in the early 1980's. Since then, SR has been widely exploited in physics, chemistry and biological sciences, and a third generation of synchrotron sources is coming into action. SR offers many new possibilities in crystallography; development for macromolecular crystallography and physics-based crystallography has led the way, but this account is concerned primarily with the potential of SR for structural chemistry.
The chemical crystallographer would probably like simply to put his diffractometer up against this intense source and get on with measurements, but it is not as simple as that! At every synchrotron source there is a large group of people involved in the development, installation, and maintenance of all the necessary instrumentation. For the source itself this includes vacuum technology, magnets and their cooling systems, electron beam injection, steering and control, monitoring of electron beam, etc. For each beamline it also includes optics, i.e. monochromators and focusing mirrors, safety systems with extensive interlocks, remote control for all alignment and other operations, detectors.
Powder Diffraction
The high intensity and low beam divergence of synchrotron radiation have allowed the design of powder diffractometers which give greatly enhanced resolution compared with conventional experimental arrangements. The resulting diffraction patterns can be analyzed to give theta angles accurate to 0.01° and intensities. As a result the indexing of a powder diffraction pattern, and the derivation of unit cell have become practicable for larger cells and cells of lower symmetry than hitherto. In favorable cases the space group and then a structural model can be derived by direct methods or by a Patterson series, using the intensities of the resolved reflections. Once a structural model has been found, refinement by the Rietveld method can use the whole diffraction pattern to improve this model, so that those regions where the peaks overlap are used as well as the fully resolved peaks. The structure determination of the clathrasil, sigma-2, by McCusker (J. Appl. Cryst. 21, 305-310) provides an excellent example of ab initio structure determination and refinement, of a compound of some complexity, for which suitable single crystals were not available. Nelmes and colleagues have used powder diffraction data for the exploration of structure at pressures much higher than atmospheric for a variety of simple and potentially important compounds, and Cheetham and Wilkinson (Angew. Chem. Int. Edn. 1992, 31, 1557-1570) have reviewed a variety of other applications of powder diffraction.In an alternative approach, "energy dispersive diffraction", or EDD, the detector is at a fixed angle and is able to resolve energies (or wavelengths). This arrangement is particularly useful when the temperature or pressure are to be varied or for time-resolved studies. For example, EDD was used at the SRS, Daresbury to observe changes in the structure of solid C70 at pressures between atmospheric and 25 GPa.
Single Crystal Diffraction with Monochromatic Radiation
The high intensity of SR makes its use for very small crystals an obvious application (Table 1a). Schulz and co-workers in their pioneering study of very small CaF2 crystals were concerned to establish vibration parameters of Ca and F in crystals where extinction effects were reduced or eliminated. For other crystals in Table 1a the aim has been structure determination, the establishment of chemical connectivity and/or constitution and stereochemistry; unit cells and structure were unknown before the experiments were done. The crystals are larger than the first group, but the unit cells are more complex, and so the average diffracted intensity is much lower.
Rubredoxin is a representative of the very large number of macromolecular crystals that have been studied, and in this area SR data has already made an enormous contribution to structural knowledge. The high intensity and low beam divergence have led directly to more and more accurate intensity data, and consequently much clearer and more interpretable electron density maps. The other examples in this table show where this improvement in data quality and quantity has been used for chemical or "small molecule" problems. In Coppens' pioneering study of a chromium complex, a very short wavelength, 0.30 Å, was chosen to minimize extinction and absorption. More recently Maslen has mapped deformation density in a variety of other compounds. In Fig. 1 deformation densities calculated from MoKα data and SR data are compared; in the latter the features and background appear very much cleaner.
With SR, the wavelength can be selected using a monochromator; the present practicable wavelength range lies between ca 0.5 and 2.0 Å. Almost all elements heavier than Mn have an absorption edge within this range. Anomalous dispersion has been widely used in macromolecular crystallography for phase determination, and to a much smaller extent with simpler structures, for example, for distinguishing neighboring elements.
The SR Laue Method
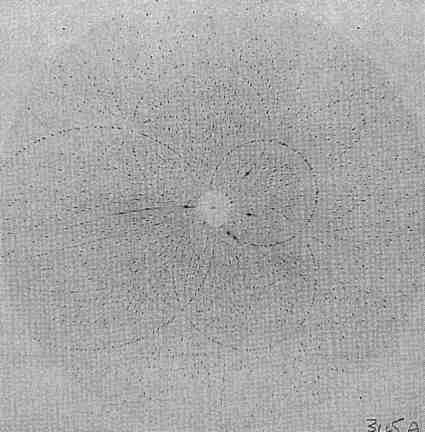 Fig. 2 Laue diffraction pattern of a complex of ably oriented lanthanum and macrocyclic ligand. The crystal is orthorhombic, with cell dimensions ca 20 Å crystal film distance 62 mm, exposure time 0.2 s on the SRS wiggler beamline at Daresbury Laboratory.
Fig. 2 Laue diffraction pattern of a complex of ably oriented lanthanum and macrocyclic ligand. The crystal is orthorhombic, with cell dimensions ca 20 Å crystal film distance 62 mm, exposure time 0.2 s on the SRS wiggler beamline at Daresbury Laboratory.A Laue diffraction pattern is shown in Fig. 2. To predict the spot positions in the pattern it is necessary to know the crystal cell and orientation, λmax and λmin (which are not, in the real experimental conditions, sharp cutoffs), and dmin= 1/2θmax for the crystal (again not a sharp cutoff). A substantial fraction of the unique reflections can be recorded on one Laue diffraction image from a suitably oriented crystal, for example, 45% for an orthorhombic crystal, nearly 90% for a tetragonal crystal (4/mmm). The Daresbury Software Suite for processing the digitized Laue film images to yield reflection intensities is widely used. Merging R factors of 5-10%, and comparisons with monochromatic diffractometer data for several proteins, show that the Laue method, when carefully used, can give good quality diffraction data. Gomez de Anderez et al. (Acta Cryst. B 1989, 45, 482-488) showed that the structure of a modest sized organic compound, C25H20N2O2 in space group P2121, could be solved and refined with Laue data; the hydrogen atoms could be located from a difference map and the final R factor was 0.053 for 1914 (single) reflections with I > 2σ(I). Other structures solved entirely from Laue data give further evidence that the accuracy of the intensity measurements is satisfactory.
Considerable effort has recently been given to deconvoluting the intensities measured for the multiple Laue spots, to give the individual components (Hao et al., Acta Cryst. A 1993, 49, 528-531). These components are predominantly low order reflections and reflections in special planes and lines in the reciprocal lattice, for example, h00, hk0, or hhh reflections. This deconvolution is important if direct method solution is to be successful with big molecules. A procedure has been developed for determining unit cell dimensions from Laue diffraction patterns. The unit cell angles and the relative values of a, b, and c can be found by converting a Laue diffraction pattern to the form known as a gnomonic projection. To obtain a, b and c on an absolute scale an attenuator with a suitable absorption edge, for example, Pd, is placed in the incident beam; λmin becomes a sharp limit and can be identified and used to give the required scaling factor (Carr et al., J. Appl. Cryst. 26, 384-387).
Laue data have been recorded for a number of crystals to test the validity of the experimental method and the software. The greatest potential of the Laue method is for time-resolved experiments, and for experiments in special environments at high temperature and/or pressure; it may also have uses for small crystals, or for crystals with a limited lifetime.
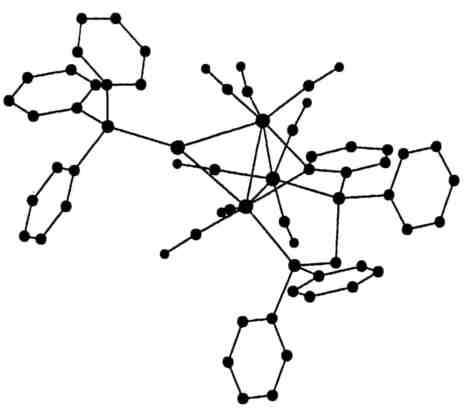 The structure of the organometallic cation, AuOs3H(CO)8{Ph2PCH2P(Ph)C6H4}(PPh3) established entirely from Laue diffraction photographs of a small crystal of its hexafluorophosphate.
The structure of the organometallic cation, AuOs3H(CO)8{Ph2PCH2P(Ph)C6H4}(PPh3) established entirely from Laue diffraction photographs of a small crystal of its hexafluorophosphate.The structure of a complex molybdosulphate was established with intensity data from seven Laue film packs (Maginn et al., Acta Cryst. B 1993, 49, 520-524). For another small organometallic crystal, Laue diffraction techniques including anomalous dispersion data established chemical constitution with R=0.078 and wR2=0.19 for 10,625 intensities with wavelengths in the range 0.24-0.65 Å (Dodd et al., Acta Cryst B 1994, 50). Anomalous dispersion effects permitted enantiomorph determination for a noncentrosymmetric but normal sized crystal of a known compound, RuC30H32P2O6 (R=0.075 for 7241 intensities).
Laue diffraction patterns have been recorded for inorganic crystals of dimensions 100 x 20 x 10 μm, and 20 x 20 x 5 μm. Ohsumi et al. (J. Appl. Cryst. 1991, 24, 340-348) chose the Laue method for their experiments at Photon Factory, Japan, to detect the diffraction patterns of minute spheres of molybdenum particles, 0.27 μm3 in volume.
Thus SR has already allowed the investigation of a variety of problems in structural chemistry which were not practicable with conventional radiation sources, structures determined for small crystals and crystalline powders, or fine details revealed by data of particularly high accuracy, or structural information from samples at high temperatures and/or pressures. Some time-resolved experiments have been done with powders and some with protein crystals. New synchrotron sources and instrumentation are being developed. We can expect that synchrotron radiation experiments will provide a rich source of new information for structural chemistry in the future.
Full paper to be published in Acta Cryst. B51, (1995).
Marjorie M. HardingLiverpool University, UK
Table 1. Examples of the use of SR in single crystal studies
| Size/μm3 | Objective | Wavelength/Å | Location | Reference | |
|---|---|---|---|---|---|
| fluorite, CaF2 | 63 | extinction free data B values | 0.91 | Doris, Hamburg | a |
| fluorite, CaF2 | 2.23 | feasibility study | 1.56 | CHESS, Cornell | b |
| aluminophosphate, AlPO4-CHA Al3P3O12-;C4H10NO+ | 35x20x15 | structure determination | 0.9 | SRS, Daresbury | c |
| organometallic gold cluster Au10(PPh3)7(S2C2(CN)2)2 | 35x10x10 | structure determination | 0.9 | SRS | d |
| lipid model compound NH4+C18H32NO6- | 280x80x16 | structure determination | 0.46 | Doris | e |
| Objective | Wavelength/Å | Location | Reference | ||
| Cr(NH3)6(CN)6 | deformation density study (first with SR) | 0.30 | CHESS | f | |
| K2PdCl4 | deformation density study | 0.70, 090 | Photon Factory, Japan | g | |
| rubredoxin, small protein 400 non-H atoms + FeS4 | 1 Å resolution data for structure refinement (previous best 1.5 Å) | 0.65 | Doris | h | |
| dodecasil 1H(SiO2)n | accurate data to resolve details of disordered sites | 0.79 | Doris | j | |
| margarite | measure weak reflections: resolve space group ambiguity | 0.5, 0.60 | Doris | k | |
References
a. R. Bachmann, H. Kohler, H. Schulz and H. P. Weber (1985) Acta Cryst. A41, 35-40.
b. W. Rieck, H. Euler, H. Schulz and W. Schildramp (1988) Acta Cryst. A44, 1099-1101.
c. M. M. Harding and B. M. Kariuki (1994) Acta Cryst. C50, in press.
d. A. K. Cheetham and A.P. Wilkinson (1992) Angew. Chem. Int. Edn. 31, 1557-1570.
e. C. W. Lehmann, J. Buschmann, P. Luger, C. Demoulin, J. H. Fuhrhop and K. Eichorn (1990) Acta Cryst. B46, 646-650.
f. F. S. Nielsen, P. Lee and P. Coppens (1986) Acta Cryst. B42, 359.
g. J. R. Hester, E. N. Maslen, N. Spadaccini, N. Ishizawa and Y. Satow (1993) Acta Cryst. B49, 842-846.
h. Z. Dauter, L. C. Sieker and K. S. Wilson (1992) Acta Cryst. B48, 42-59.
j. G. Miehe, T. Vogt, H. Fuess and U. Muller (1993) Acta Cryst. B49, 745-754.
k. D. Kassner, W. H. Baur, W. Joswig, K. Eichorn, M. Wendschuh-Josties and V. Kupcik (1993) Acta Cryst. B49, 646-654.
